
Abstract
Computational methods and recently modern machine learning methods have played a key role in structure-based drug design. Though several benchmarking datasets are available for machine learning applications in virtual screening, accurate prediction of binding affinity for a protein-ligand complex remains a major challenge. New datasets that allow for the development of models for predicting binding affinities better than the state-of-the-art scoring functions are important. For the first time, we have developed a dataset, PLAS-5k comprised of 5000 protein-ligand complexes chosen from PDB database. The dataset consists of binding affinities along with energy components like electrostatic, van der Waals, polar and non-polar solvation energy calculated from molecular dynamics simulations using MMPBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method. The calculated binding affinities outperformed docking scores and showed a good correlation with the available experimental values. The availability of energy components may enable optimization of desired components during machine learning-based drug design. Further, OnionNet model has been retrained on PLAS-5k dataset and is provided as a baseline for the prediction of binding affinities.
Measurement(s) | Binding Affinity |
Technology Type(s) | Molecular dynamics simulation/MM-PBSA |
Factor Type(s) | 3D-protein structures |
Sample Characteristic - Organism | NA |
Sample Characteristic - Environment | NA |
Sample Characteristic - Location | NA |
Similar content being viewed by others
Background & Summary
The task of predicting binding affinity of a protein-ligand (PL) complex is of cardinal significance in the drug design pipeline1. In general, determining the binding affinities of PL complex through experimental assays is laborious and economically non-viable. To mitigate the investments in drug discovery, in-silico methods have been adopted over traditional experiments in initial stages of drug design. Experimentally inaccessible molecular interactions and mechanisms can be studied through computational methods. Computer-aided drug design (CADD) is one such promising area of drug discovery and helps to predict the best interaction model between a PL and use scoring functions to estimate the strength of the binding. In recent decades, researchers have increasingly recognized that molecular dynamics simulation (MD) helps to overcome the major limitations of docking calculations that do not sample protein conformational rearrangements during the ligand-binding process. MD simulations based on binding affinity calculations using molecular mechanics with Poisson-Boltzmann (MM-PBSA/MM-GBSA) are therefore expected to provide significant contributions to real-world problems such as identification of hit and lead optimization. The most important post-processing methods for calculating the binding free energy of a PL complex include molecular mechanics with Poisson-Boltzmann/Generalized-Born and surface area (MM-PBSA/MM-GBSA), and alchemical approaches like thermodynamic integration and free-energy perturbation (FEP)2. Apart from these methods, machine learning (ML) models have also been used for binding affinity predictions (BAP)3. ML models can enhance data-driven decision-making and have the potential to speed up the drug discovery process. The current ML models developed for BAP are grouped by the different types of encoding, topology, and atom pairs.
Interaction fingerprints framework used for binding site comparison has proven to be successful in many applications, ranging from assessment of docking poses to the evaluation of novel PL complexes4. Some of the applications include structural Protein-Ligand interaction fingerprint5, Protein-ligand extended connectivity fingerprint6 and most recently Substructural Molecular and Protein-Ligand Interaction Pattern Score7. In 3D grid-based studies, PL complex is represented using a 3D grid representation. AtomNet was one of the first published models that used a convolutional neural network for affinity prediction8. Few other models include KDEEP9, Pafnucy10, DeepAtom11, and BindScope12.
Another deep learning method that could reach the state-of-the-art performance in predicting PL interaction is graph neural network. Few applications include GraphBAR13, structure-aware interactive graph neural network14, the model developed by Lim et al.15, and PotentialNet16. Apart from these models, other models such as MathDL17 and TopologyNet18 encode interactions PL using methods from algebraic topology. Models such as DeepBindRG19, DeepVS20, and OnionNet21 are focused on interacting atom environments of complex structures.
A number of datasets facilitate the development of ML-based scoring functions22 for BAP. Such ML scoring functions use PL information either as a complex or as two different entities. Several benchmarking datasets are publicly available. The BindingMOAD23, PDBbind24, and CSAR datasets25 were compiled to aid in the prediction of binding affinities based on experimental PL complex structures. The KIBA26 and DAVIS27 dataset highlights the bioactivities of the kinase protein family and their relevant inhibitors and does not include the structural information of PL complexes. The DUD and DUD-E datasets28 were designed to evaluate docking enrichment performance. However, the existing datasets are limited to crystal structures of PL complex despite the widely accepted role of protein flexibility in molecular recognition29. This simplified description of the complex narrows down the accuracy of the binding pose prediction and their corresponding scoring functions30. Herein, MD simulations play a major role in capturing the conformational changes in the complex structure thereby helping in the accurate prediction of binding affinity. This could also improve the size of the diverse datasets and enhance the existing scoring functions based on energetic contributions to binding affinities. In existing datasets, energy components are unavailable, although they are highly important for lead optimization and target-specific drug design. MM-PBSA is a method that provides individual energy components along with the overall binding affinities from MD trajectories. In recent years, MM-PBSA has become a popular method to estimate the ligand binding affinities and it has several applications31. Few examples include, development of potential anticancer compounds31,32, understanding resistance mechanism of drugs33, neural disorder34, blood disorder35, immune disorder36, inflammatory disorder37, metabolic disorder38, and many other major diseases39,40. Apart from these PL interactions, MM-PBSA calculations also play a major role in other biomolecular studies such as protein folding, protein-protein interaction41, and others42. Various studies also highlight the successful applications of MM-PBSA in virtual screening for identification of potential lead compounds43. The most recent application includes identification of suitable inhibitors for COVID-19 targets and also repurposing of existing FDA approved drugs44.
In this work, we employed MD simulations on 5000 PL complexes to calculate the binding affinities using MM-PBSA approach. To best of our knowledge, this is the first MD-based dataset that provides binding affinities along with non-covalent interaction components. Comparisons have been made by calculating the correlation coefficients between experimentally determined values to that of calculated affinities (MM-PBSA and Docking). As a baseline, we have trained the OnionNet framework on our dataset. We believe that PLAS-5k and further work in this direction will provide the necessary impetus for the development of data-driven methods for drug design tasks such as hit identification, lead optimization, de novo molecular design, etc.
Methods
Data curation
In this article, as a first step towards the development of dataset, we have selected 5000 complexes randomly from PDB23 based on the following criteria (i) In these complexes, ligand is chosen to be either a small organic molecule or a peptide, (ii) the complex structures within 2.5 Å resolution.
System preparation
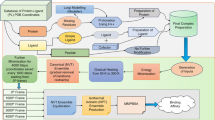
Each protein-ligand complex chosen is composed of protein, ligand, cofactor(s) and crystal water molecules. The procedure of preparing the complexes for MD simulations is discussed in detail in the following sections, and is shown in Fig. 1.

Protocol for input preparation and simulations.
Protein preparation
Most of the chosen experimental protein structures are monomers, while few can be functional as multimers. In cases of multimers, the subunits within a distance of 8 Å of ligand molecule were considered for complex preparation. In case of missing residues, MODELLER program was used to build the missing residues in PDB structures as loop regions45. Further, protonation states of the residues in the protein structures were determined using the H++ server46 at the physiological pH of 7.4. For the simulations, Amber ff14SB parameters were used for proteins47.
Ligand preparation
The information of total charge on the ligand was retrieved using ligand-expo and hydrogen atoms were added to the ligand using GaussView48 in appropriate positions49. Similar procedure were adopted for the cofactors. The forcefield parameters for ligand and cofactors were obtained from General AMBER force field (GAFF2)50 using Antechamber program51 of Ambertools52,53. AM1-BCC charges were assigned to the atoms of ligand and cofactor(s). In case of peptides, Amber ff14SB47 forcefield was used.
Complex preparation
As water molecules play an important role in mediating protein-ligand interactions, the crystal waters associated with the selected subunits of proteins have been considered for the studies.The “tleap” program of AmberTools52,53 was used to generate a complex. The systems were solvated in an orthorhombic water box with a 10 Å extension from the protein. To maintain the charge neutrality of the system, counter ions (Na+ or Cl−) were added.
Simulation setup
Energy minimization
Minimization was performed in two steps. First, the protein backbone atoms were restrained using a harmonic potential with a force constant of 10 kcal/mol/Å2 in 1000 step minimization using L-BFGS minimizer was carried out. Further, the spring constant was reduced in ten steps and energy minimization was performed. In each step, the force constant was scaled by half. Finally, the harmonic restraints were turned off and minimization was carried out for another 1000 steps.
Simulating to target temperature (300 K)
After energy minimization, short MD simulation was performed with a timestep of 2 fs in NPT ensemble, with position restraints on backbone atoms using harmonic potential with spring constant of 1 kcal/mol/Å2. The particle mesh Ewald (PME) method was used to compute the long range interactions and the non-bonded interactions were truncated at 10.0 Å. The bonds involving hydrogen atoms were constrained. The temperature of the system was maintained using Langevin thermostat with a friction coefficient of 5 ps−1. The system temperature was raised from 50 K to 300 K by increasing the temperature by 1 K in every 100 steps (200 fs). Finally, after reaching target temperature (300 K), simulations were performed for 1 ns in the NVT ensemble.
Multiple independent simulations
Studies have reported that many short run independent simulations are more effective than a single long run, and it will decrease the uncertainty for the predicted binding affinities54,55,56. In general, the independent simulations are performed with different set of random initial velocities and initial structures taken during the minimization. The initial structures were generated from energy minimization in 40000 steps. At every 10000 steps, the structures were saved to start five independent simulations (including the starting structure).
In the next stage, all the restraints were released and the atoms were allowed to move freely. The system was equlibrated in the NPT ensemble at 300 K and 1 atm using a Langevin thermostat and Monte Carlo barostat for 2 ns. Finally, a production run was performed for 4 ns in the NPT ensemble, and the trajectories were saved every 100 ps for the post-processing analysis and free energy calculations. Molecular dynamics simulations have been carried out using the OpenMM 7.2.0 program57.
Molecular-Mechanics Poisson Boltzmann Surface Area (MM-PBSA) calculations
MM-PBSA has been extensively used in CADD, as it is less expensive compared to alchemical free energy methods. Binding free energy of a PL complex is calculated according to the following equation.
Further, ΔEMM is divided into sum of electrostatic interaction energy ΔEele, and van der Waals interaction energy ΔEvdw (Eq. (2)). The solvation free energy ΔGsol, is defined as sum of polar ΔGpol, and non-polar contributions ΔGnp (Eq. (3)).
Polar solvation energy, ΔGpol was calculated using the PBSA method as implemented in the AMBER20 program and non-polar contributions were determined using Linear Combinations of Pairwise Overlap (LCPO) method58.
Both experimental and CADD have highlighted the role of water molecules in PL binding as they aid in water mediated hydrogen bond interactions59,60,61. In our study we have considered two water molecules (see SI for more details and Supplementary Figure S1), which are near to the PL interaction site. The internal dielectric constant 4 was considered, as several studies reported good performance in predicting binding affinity62,63,64. The binding affinity for each complex was calculated by single trajectory approach. From the complex, protein and ligand are extracted and their affinities were calculated separately. The reported binding affinities are the mean of the ΔG calculated from all the five independent runs.
Docking protocol
In structure-based drug design, docking studies have been used to determine the binding pose and affinities. The docking results are obtained by the simplified description of the complex which lacks true dynamics of the system and explicit water molecules30. On the other hand, it is been reported that end-point methods, such as MM-PBSA/MM-GBSA, are based on snapshots of MD simulations trajectories and they tend to overcome the limitations of docking and provide more accurate results than docking scoring functions. In this work, docking studies were performed for structures with known experimental binding affinities using AutoDock vina65. The crystal structures of all PL complexes were retrieved from PDB database and were refined by removing heteroatoms. Further, hydrogen atoms were added and Kollman charges were assigned to the protein structures. For ligands, Gasteiger partial atomic charges were assigned and all flexible torsion angles were defined using AUTOTORS. The active site of each target was discretized through a grid and the docking calculations were performed with default parameters66.
Data Records
PLAS-5k dataset (https://hai.iiit.ac.in/datasets.html) can be searched using the PDB id as a query and an example of data retrieval from the PLAS-5k database is illustrated in Supplementary Figure S2. After submitting the query the results are displayed and it gives information on the total binding affinity and different energy components like van der Waals interaction energy, electrostatic energy, polar and non-polar solvation energies. Structural visualization of the protein-ligand complex is available for each entry. The initial structures of all the 5000 protein ligand complexes are available in PDB format and the csv file containing information about binding affinity components can be accessed through figshare67.
Technical Validation
Overall structures of the protein-ligand complexes
In the present work, we performed MD simulations to capture several conformations of the PL complex to incorporate the flexibility of protein in binding affinity calculations. The experimental structure of a complex is taken as a reference in the RMSD calculation of both protein and ligand over the simulation trajectory. In order to capture the conformations of ligand, the structure of the protein was superimposed primarily and the RMSD of protein and ligand was measured separately for all five independent runs. The cumulative RMSD of protein and ligand for each of the complexes is calculated over all 200 frames (40 from each simulation), and the corresponding distributions are shown in Supplementary Figure S3. The long tail in the distributions are due to the presence of flexible groups present in protein (loops) and ligand. Since the RMSD for ligands peak at <1 Å and the majority fall below 3 Å, the ligands remain stably bound throughout the simulations. Our dataset covers wide range of ligands and the distribution of molecular weights of these ligands is shown in Supplementary Figure S4.
Experimentally, the binding affinity of a protein-ligand complex is expressed in terms of dissociation constant (Kd) or inhibition constant (Ki). This experimentally determined binding equilibrium constant is related to the binding free energy as,
MM-PBSA approach has been widely accepted as an efficient and reliable free energy method in estimating PL binding interactions and has high correlation with experimental binding affinity68 especially for a given protein with respect to multiple ligands. A combination of interaction energetic components from MM-PBSA and ML methods help in developing models that could identify suitable inhibitors for a specific target69. The calculated binding free energies using MM-PBSA method span a wide range of values capturing a broad distribution suitable for developing ML models (Supplementary Figure S5). Having a knowledge on these large interval values of calculated binding affinity for diverse dataset, would help in extracting feature representation of PL complexes and train reliable regression models that can help in predicting binding affinity of a novel complex, and for use in other applications such as molecule generation.
Comparison of experimental vs calculated binding affinities
For comparison study, we made a subset (2000 complexes) of 5000 complexes, whose experimental binding affinities are known. The calculated binding affinities based on docking studies and MM-PBSA method were compared with the experimental values. The Spearman rank correlation coefficient (Rs) and Pearson correlation coefficient (Rp) were used to evaluate the ranking of binding affinities and their correlation with experimental data respectively. As seen in Fig. 2, the (Rp) was 0.385 for docking studies with (Rs) of 0.390, while the studies based on MM-PBSA show relatively stronger correlation with (Rp) and (Rs) of 0.585 and 0.598 respectively. This indicates that ML based scoring functions developed using PLAS-5k dataset are expected to be more reliable than the traditional scoring functions.

Correlation plots between the experimental and calculated binding affinities for a subset with 2000 pdbids. The binding affinities are calculated (a) using Auto-dock Vina, and (b) using MM-PBSA.
Class specific performance
The dataset was classified into seven different classes as follows: (i) Transferases, (ii) Hydrolases, (iii) Isomerases, (iv) Oxido-reductases, (v) Ligases, (vi) Lyases, and (vii) Others. These enzymes are essential biological catalysts involved in a number of chemical transformations pertaining to life. From the Table 1 and Supplementary Figures S6, S7 it can be noted that the binding affinities predicted through MM-PBSA shows good correlation with the experimental value for most of the classes compared to docking affinities.
Target-specific performance: experimental vs docking and MM-PBSA
Performance of HIV-1 protease targets
HIV-1 Protease is an essential enzyme in the life cycle of HIV as they play an important role in viral replication and maturation. The discovery of HIV-1 protease inhibitors in the last 25 years is a major success in structure based drug design. There are totally nine FDA approved protease inhibitors. A lot of efforts have been made in drug discovery process in development of next-generation protease inhibitors beyond the currently approved protease inhibitors. This shows that until today, HIV-1 protease continues to be one of the attractive targets as they continue to play an important role in drug discovery70,71,72,73,74.
Docking studies of HIV-1 protease with FDA approved drugs shows that (Rp) and (Rs) were 0.25 and 0.09 respectively (Fig. 3a). As shown in Fig. 3b, in case of MM-PBSA calculations, the simulation results show good correlation of 0.52 (Rp) and 0.68 (Rs). The linear correlation coefficient (Rp) is marginally good, but the Spearman ranking coefficient showed better performance than that of Rp, which is more essential characteristic in drug discovery.

Prediction of binding affinity based on correlation with experimental data: FDA approved drugs for HIV-I protease targets (a) Experimental vs Docking, (b) Experimental vs MM-PBSA; For Tuberculosis targets - (c) Experimental vs Docking (d) Experimental vs MM-PBSA.
Performance of tuberculosis targets
Tuberculosis (TB), a contagious and potentially fatal disease continues to be a major health problem worldwide. Though tremendous progress has been made in anti-TB therapy over the last seven decades to eradicate the disease, TB continues to affect millions of people worldwide75. Numerous efforts have been made in drug discovery to search new antitubercular agents that can inhibit the drug resistant strains76,77. With this motivation, we selected TB targets to assess the performance of our dataset. As observed for HIV-1 protease, even the TB targets showed better performance in case of MM-PBSA calculations with correlation values (Rp) and (Rs) ranking of 0.56 and 0.49 respectively, whereas the docking results showed values of 0.24 (Rp) and 0.28 (Rs). The correlation plots for tuberculosis targets are shown in Fig. 3(c,d).
Components of the binding free energies
Non-bonded/non-covalent interactions play a crucial role in stabilizing the protein-ligand complexes and a detailed understanding of these interactions can provide valuable insights in drug design. One of the advantages about PLAS-5k is that it provides protein-ligand interactions in terms of electrostatic interactions, van der Waals interactions, polar and non-polar contributions to solvation free energy. The distribution plots are shown in Supplementary Figure S8. A knowledge of these individual energy components (Eq. (1)) could help the researchers to have an tailored procedure in lead optimization of drug discovery process.
Machine learning benchmark
Prediction of binding affinity of a PL complex is a critical step in drug design, and ML methods have begin to make significant contributions. One of the pioneering model is OnionNet21. Taking various features derived from 3D molecular structure as a input and known binding affinities it predicts binding affinity for a unknown complex via use of Convolutional Neural Network (CNN). PLAS-5k data, was trained and tested using OnionNet model. A 10-fold validation was employed, where the dataset was divided into 10 equal parts and 9-parts were used for training the model, rest for testing. This was employed due to the size constraint of the dataset. The average RMSE across all the 10-fold split was 5.7 kcal/mol and with an Rp of 0.96, as shown in Fig. 4.

Pearson correlation coefficient after training OnionNet on PLAS-5k database.
Code availability
No custom code was used in the creation of this database. We used OnionNet21 http://github.com/zhenglz/onionnet/ ML model to train on PLAS-5k dataset. Ambertools52, GaussView48, MODELLER45, and H++ server46 were used for preparation of complex containing protein, ligand, and cofactor(s). MD simulations were carried using OpenMM 7.2.0 program57.
References
Kairys, V., Baranauskiene, L., Kazlauskiene, M., Matulis, D. & Kazlauskas, E. Binding affinity in drug design: experimental and computational techniques. Expert opinion on drug discovery 14, 755–768 (2019).
Srivastava, H. K. & Sastry, G. N. Molecular dynamics investigation on a series of hiv protease inhibitors: assessing the performance of mm-pbsa and mm-gbsa approaches. Journal of chemical information and modeling 52, 3088–3098 (2012).
Kimber, T. B., Chen, Y. & Volkamer, A. Deep learning in virtual screening: Recent applications and developments. International Journal of Molecular Sciences 22, 4435 (2021).
Mordalski, S., Kosciolek, T., Kristiansen, K., Sylte, I. & Bojarski, A. J. Protein binding site analysis by means of structural interaction fingerprint patterns. Bioorganic & medicinal chemistry letters 21, 6816–6819 (2011).
Da, C. & Kireev, D. Structural protein–ligand interaction fingerprints (splif) for structure-based virtual screening: method and benchmark study. Journal of chemical information and modeling 54, 2555–2561 (2014).
Wójcikowski, M., Kukiełka, M., Stepniewska-Dziubinska, M. M. & Siedlecki, P. Development of a protein–ligand extended connectivity (plec) fingerprint and its application for binding affinity predictions. Bioinformatics 35, 1334–1341 (2019).
Kumar, S. & Kim, M.-H. Smplip-score: predicting ligand binding affinity from simple and interpretable on-the-fly interaction fingerprint pattern descriptors. Journal of cheminformatics 13, 1–17 (2021).
Wallach, I., Dzamba, M. & Heifets, A. Atomnet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. (2015).
Jiménez, J., Skalic, M., Martinez-Rosell, G. & De Fabritiis, G. K deep: protein–ligand absolute binding affinity prediction via 3d-convolutional neural networks. Journal of chemical information and modeling 58, 287–296 (2018).
Stepniewska-Dziubinska, M. M., Zielenkiewicz, P. & Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 34, 3666–3674 (2018).
Li, Y., Rezaei, M. A., Li, C. & Li, X. Deepatom: a framework for protein-ligand binding affinity prediction. In 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 303–310 (IEEE, 2019).
Skalic, M., Martnez-Rosell, G., Jiménez, J. & De Fabritiis, G. Playmolecule bindscope: large scale cnn-based virtual screening on the web. Bioinformatics 35, 1237–1238 (2019).
Son, J. & Kim, D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PloS one 16, e0249404 (2021).
Li, S. et al. Structure-aware interactive graph neural networks for the prediction of protein-ligand binding affinity. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, 975–985 (2021).
Lim, J. et al. Predicting drug–target interaction using a novel graph neural network with 3d structure-embedded graph representation. Journal of chemical information and modeling 59, 3981–3988 (2019).
Feinberg, E. N. et al. Potentialnet for molecular property prediction. ACS central science 4, 1520–1530 (2018).
Nguyen, D. D., Gao, K., Wang, M. & Wei, G.-W. Mathdl: mathematical deep learning for d3r grand challenge 4. Journal of computer-aided molecular design 34, 131–147 (2020).
Cang, Z. & Wei, G.-W. Topologynet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLoS computational biology 13, e1005690 (2017).
Zhang, H., Liao, L., Saravanan, K. M., Yin, P. & Wei, Y. Deepbindrg: a deep learning based method for estimating effective protein–ligand affinity. PeerJ 7, e7362 (2019).
Pereira, J. C., Caffarena, E. R. & Dos Santos, C. N. Boosting docking-based virtual screening with deep learning. Journal of chemical information and modeling 56, 2495–2506 (2016).
Zheng, L., Fan, J. & Mu, Y. Onionnet: a multiple-layer intermolecular-contact-based convolutional neural network for protein–ligand binding affinity prediction. ACS omega 4, 15956–15965 (2019).
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M. & Taranto, A. G. Structure-based virtual screening: from classical to artificial intelligence. Frontiers in chemistry 8, 343 (2020).
Hu, L., Benson, M. L., Smith, R. D., Lerner, M. G. & Carlson, H. A. Binding moad (mother of all databases). Proteins: Structure, Function, and Bioinformatics 60, 333–340 (2005).
Wang, R., Fang, X., Lu, Y., Yang, C.-Y. & Wang, S. The pdbbind database: methodologies and updates. Journal of medicinal chemistry 48, 4111–4119 (2005).
Dunbar, J. B. Jr et al. Csar data set release 2012: ligands, affinities, complexes, and docking decoys. Journal of chemical information and modeling 53, 1842–1852 (2013).
Tang, J. et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. Journal of Chemical Information and Modeling 54, 735–743 (2014).
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology 29, 1046–1051 (2011).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (dud-e): better ligands and decoys for better benchmarking. Journal of medicinal chemistry 55, 6582–6594 (2012).
Amaral, M. et al. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nature communications 8, 1–14 (2017).
Pantsar, T. & Poso, A. Binding affinity via docking: fact and fiction. Molecules 23, 1899 (2018).
Liu, H., Han, R., Li, J., Liu, H. & Zheng, L. Molecular mechanism of r-bicalutamide switching from androgen receptor antagonist to agonist induced by amino acid mutations using molecular dynamics simulations and free energy calculation. Journal of computer-aided molecular design 30, 1189–1200 (2016).
Yang, X. et al. Docking and molecular dynamics studies on triclosan derivatives binding to fabi. Journal of molecular modeling 23, 25 (2017).
Li, D., Zhang, Y., Zhao, R.-N., Fan, S. & Han, J.-G. Investigation on the mechanism for the binding and drug resistance of wild type and mutations of g86 residue in hiv-1 protease complexed with darunavir by molecular dynamic simulation and free energy calculation. Journal of molecular modeling 20, 1–11 (2014).
Ekhteiari Salmas, R. et al. Biological insights of the dopaminergic stabilizer acr16 at the binding pocket of dopamine d2 receptor. ACS chemical neuroscience 8, 826–836 (2017).
Kragh-Hansen, U. et al. Mutants and molecular dockings reveal that the primary l-thyroxine binding site in human serum albumin is not the one which can cause familial dysalbuminemic hyperthyroxinemia. Biochimica et Biophysica Acta (BBA)-General Subjects 1860, 648–660 (2016).
Verma, R. et al. Probing binding mechanism of interleukin-6 and olokizumab: in silico design of potential lead antibodies for autoimmune and inflammatory diseases. Journal of Receptors and Signal Transduction 36, 601–616 (2016).
Chaudhary, N. & Aparoy, P. Deciphering the mechanism behind the varied binding activities of coxibs through molecular dynamic simulations, mm-pbsa binding energy calculations and per-residue energy decomposition studies. Journal of Biomolecular Structure and Dynamics 35, 868–882 (2017).
Qian, H., Chen, J., Pan, Y. & Chen, J. Molecular modeling studies of 11β-hydroxysteroid dehydrogenase type 1 inhibitors through receptor-based 3d-qsar and molecular dynamics simulations. Molecules 21, 1222 (2016).
Begum, J. et al. An evaluation of indirubin analogues as phosphorylase kinase inhibitors. Journal of Molecular Graphics and Modelling 61, 231–242 (2015).
Tzoupis, H. et al. Elucidation of the binding mechanism of renin using a wide array of computational techniques and biological assays. Journal of Molecular Graphics and Modelling 62, 138–149 (2015).
Wang, L. et al. Discovery and identification of cdc37-derived peptides targeting the hsp90–cdc37 protein–protein interaction. RSC advances 5, 96138–96145 (2015).
Wang, C., Greene, D., Xiao, L., Qi, R. & Luo, R. Recent developments and applications of the mmpbsa method. Frontiers in molecular biosciences 4, 87 (2018).
Poli, G., Granchi, C., Rizzolio, F. & Tuccinardi, T. Application of mm-pbsa methods in virtual screening. Molecules 25, 1971 (2020).
Chowdhury, K. H. et al. Drug repurposing approach against novel coronavirus disease (covid-19) through virtual screening targeting sars-cov-2 main protease. Biology 10, 2 (2021).
Pettersen, E. F. et al. Ucsf chimeraΓÇöa visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612 (2004).
Gordon, J. C. et al. H++: a server for estimating p k as and adding missing hydrogens to macromolecules. Nucleic acids research 33, W368–W371 (2005).
Maier, J. A. et al. ff14sb: improving the accuracy of protein side chain and backbone parameters from ff99sb. Journal of chemical theory and computation 11, 3696–3713 (2015).
Dennington, R., et al. Gaussview, version 5 (2009).
Feng, Z. et al. Ligand depot: a data warehouse for ligands bound to macromolecules. Bioinformatics 20, 2153–2155 (2004).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. Journal of computational chemistry 25, 1157–1174 (2004).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Automatic atom type and bond type perception in molecular mechanical calculations. Journal of molecular graphics and modelling 25, 247–260 (2006).
Case, D. A. et al. The amber biomolecular simulation programs. Journal of computational chemistry 26, 1668–1688 (2005).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science 3, 198–210 (2013).
Adler, M. & Beroza, P. Improved ligand binding energies derived from molecular dynamics: replicate sampling enhances the search of conformational space. Journal of chemical information and modeling 53, 2065–2072 (2013).
Wright, D. W., Hall, B. A., Kenway, O. A., Jha, S. & Coveney, P. V. Computing clinically relevant binding free energies of hiv-1 protease inhibitors. Journal of chemical theory and computation 10, 1228–1241 (2014).
Sadiq, S. K., Wright, D. W., Kenway, O. A. & Coveney, P. V. Accurate ensemble molecular dynamics binding free energy ranking of multidrug-resistant hiv-1 proteases. Journal of chemical information and modeling 50, 890–905 (2010).
Eastman, P. et al. Openmm 7: Rapid development of high performance algorithms for molecular dynamics. PLoS computational biology 13, e1005659 (2017).
Miller, B. R. III et al. Mmpbsa. py: an efficient program for end-state free energy calculations. Journal of chemical theory and computation 8, 3314–3321 (2012).
Aldeghi, M., Bodkin, M. J., Knapp, S. & Biggin, P. C. Statistical analysis on the performance of molecular mechanics poisson–boltzmann surface area versus absolute binding free energy calculations: Bromodomains as a case study. Journal of chemical information and modeling 57, 2203–2221 (2017).
Zhu, Y.-L., Beroza, P. & Artis, D. R. Including explicit water molecules as part of the protein structure in mm/pbsa calculations. Journal of Chemical Information and Modeling 54, 462–469 (2014).
Maffucci, I., Hu, X., Fumagalli, V. & Contini, A. An efficient implementation of the nwat-mmgbsa method to rescore docking results in medium-throughput virtual screenings. Frontiers in chemistry 6, 43 (2018).
Wright, D. W. et al. Application of esmacs binding free energy protocols to diverse datasets: Bromodomain-containing protein 4. Scientific Reports 9 (2019).
Sun, H., Li, Y., Tian, S., Xu, L. & Hou, T. Assessing the performance of mm/pbsa and mm/gbsa methods. 4. accuracies of mm/pbsa and mm/gbsa methodologies evaluated by various simulation protocols using pdbbind data set. Physical Chemistry Chemical Physics 16, 16719–16729 (2014).
Hou, T., Wang, J., Li, Y. & Wang, W. Assessing the performance of the molecular mechanics/poisson boltzmann surface area and molecular mechanics/generalized born surface area methods. ii. the accuracy of ranking poses generated from docking. Journal of computational chemistry 32, 866–877 (2011).
Trott, O. & Olson, A. J. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 31, 455–461 (2010).
Morris, G. M. et al. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. Journal of computational chemistry 30, 2785–2791 (2009).
Korlepara, D. B. et al. Plas-5k: Dataset of protein-ligand affinities from molecular dynamics for machine learning applications. Figshare https://doi.org/10.6084/m9.figshare.c.6144555.v1 (2022).
Wang, C. et al. Calculating protein–ligand binding affinities with mmpbsa: Method and error analysis. Journal of computational chemistry 37, 2436–2446 (2016).
Wang, E. et al. End-point binding free energy calculation with mm/pbsa and mm/gbsa: strategies and applications in drug design. Chemical reviews 119, 9478–9508 (2019).
Ghosh, A. K., Osswald, H. L. & Prato, G. Recent progress in the development of hiv-1 protease inhibitors for the treatment of hiv/aids. Journal of medicinal chemistry 59, 5172–5208 (2016).
Batman, G., Hampson, L. & Hampson, I. N. Lessons from repurposing hiv drugs: a prospective novel strategy for drug design. Future Virology 6, 1021–1023 (2011).
Sang, P., Tian, S.-H., Meng, Z.-H. & Yang, L.-Q. Anti-hiv drug repurposing against sars-cov-2. RSC Advances 10, 15775–15783 (2020).
Harrison, C. Coronavirus puts drug repurposing on the fast track. Nature biotechnology 38, 379–381 (2020).
Mahdi, M. et al. Analysis of the efficacy of hiv protease inhibitors against sars-cov-2′s main protease. Virology journal 17, 1–8 (2020).
Ginsberg, A. M. & Spigelman, M. Challenges in tuberculosis drug research and development. Nature medicine 13, 290–294 (2007).
Riccardi, G. & Pasca, M. R. Trends in discovery of new drugs for tuberculosis therapy. The Journal of antibiotics 67, 655–659 (2014).
Nguta, J. M., Appiah-Opong, R., Nyarko, A. K., Yeboah-Manu, D. & Addo, P. G. Current perspectives in drug discovery against tuberculosis from natural products. International Journal of Mycobacteriology 4, 165–183 (2015).
Acknowledgements
We thank Dr. Sethuraman Ramanathan and Mr. Konala Verma, Intel India, Prof. S Bapi Raju and Dr. Vinod P K, IIIT Hyderabad for fruitful discussions, Ms. Indhu Ramachandran for coordinating this project and Akshaya Karthikeyan, Manan Goel, Vijay Vignesh, Arihanth Tadanki, Karthik Viswanathan, Kanakala Ganesh Chandan and Sriram Devata for their initial involvement. We thank IHub-Data for support. The authors thank IIT Delhi and IIIT Hyderabad HPC facilities for computational resources. DN acknowledges financial support by INSPIRE faculty research grant (DST/INSPIRE/04/2018/000455) provided by Department of Science and Technology, India. UDP thanks DST-SERB (CRG/2021/008036) and Kohli Center on Intelligent Systems, IIIT Hyderabad for support.
Author information
Authors and Affiliations
Contributions
D.P. conceived the study, D.B.K. set up the protocol, and analysed the data. C.S.V. and D.B.K. contributed to the writing of the manuscript. S.R. constructed the database and visualization of the dataset. B.S. wrote code for data download. S.M. and S.R. trained ML model. V.K., C.S.V. and S.S. performed docking studies. S.S. helped in editing the manuscript. D.B.K., C.S.V., S.J., P.K.P., S.R., S.M., S.S., V.K., C.M., A.G. contributed in preparation of dataset and simulation. D.N., A.P.B. and D.P. provided administrative guidance in the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41597_2022_1631_MOESM1_ESM.pdf
Supplementary Information for: PLAS-5k: Dataset of Protein-Ligand Affinities from Molecular Dynamics for Machine Learning Applications
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korlepara, D.B., Vasavi, C.S., Jeurkar, S. et al. PLAS-5k: Dataset of Protein-Ligand Affinities from Molecular Dynamics for Machine Learning Applications. Sci Data 9, 548 (2022). https://doi.org/10.1038/s41597-022-01631-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01631-9
