
Abstract
Lonicera japonica (honeysuckle) is one of the most important medicinal plants and widely utilized in traditional Chinese medicine. At present, there are many varieties of honeysuckle used in cultivation, among which Sijihua variety are widely cultivated due to its wide adaptability, stress resistance, early flowering and high yield. In this study, we assembled the genome of Sijihua, which was approximately 886.04 Mb in size with a scaffold N50 of 79.5 Mb. 93.28% of the total assembled sequences were anchored to 9 pseudo-chromosomes by using PacBio long reads and Hi-C sequencing data. We predicted 39,320 protein-coding genes and 92.87% of them could be annotated in NR, GO, KOG, KEGG and other databases. In addition, we identified 644 tRNAs, 2,156 rRNAs, 109 miRNAs and 5,502 pseudogenes from the genome. The chromosome-scale genome of Sijihua will be a significant resource for understanding the genetic basis of high stress-resistance, which will facilitate further study of the genetic diversity and accelerate the genetic improvement and breeding of L. japonica.
Measurement(s) | Lonicera japonica • RNA sequencing • genome assembly • sequence annotation |
Technology Type(s) | SMRT Sequencing • RNA sequencing • Hi-C • biomolecular annotation design |
Factor Type(s) | Genotype |
Sample Characteristic - Organism | Lonicera japonica |
Sample Characteristic - Environment | occurrence |
Sample Characteristic - Location | Shandong Province |
Similar content being viewed by others


Background & Summary
Lonicera japonica (Caprifoliaceae), is perennial and evergreen twining vine, commonly known as Jinyinhua or Rendong, plays a very important role in traditional Chinese medicine1. Meanwhile, L. japonica has been cultivated as an ornamental plant in many areas because of its varying colors and attractive smell2. L. japonica firstly recorded as medicine can be traced back to the Jin dynasty in China, then it also recorded in ‘Ben Cao Gang Mu’ which is famous classical book of Chinese material medica1. The first place in China where L. japonica was planted massively is Fengqiu county of Henan province, and it has been used to treat exogenous wind-heat, febrile disease, sore, carbuncle, furuncle and some infectious diseases3. It was found that the extracts of L. japonica and its chemical components have a variety of pharmacological effects, including anti-inflammatory, antibacterial, antiviral, antioxidant, liver protection and anti-tumor1. As L. japonica being used and cultivated in more and more areas, its chemical constituents have been studied widely. L. japonica contains a variety of medicinal ingredients, such as flavonoids, essential oil, triterpenoid soap and organic acids4,5, which allows honeysuckle to perform many pharmacological functions.
L. japonica has always been the object of researches and many scientists mainly focused on the biosynthesis of active medicinal ingredients and differential gene expression patterns between different tissues with the help of transcriptomics6,7,8. However, with the development of sequencing technology and the reduction of sequencing cost, progressively plant genomes are available, including L. japonica, whose Institute of Medicinal Plant Development (IMPLAD) germplasm registration number is 101074289. The study about Lj10107428 provided proof for a whole genome duplication (WGD) event and showed the expression of related biosynthetic genes was correlated with the accumulation of carotenoids and suggested the role of carotenoid degradation in the dynamic coloring of L. japonica by assembling the whole genome and transcriptomic analysis9. Nevertheless, a genome of one variety is not enough to represent the genetic resources of the species. The availability of the genomes of different varieties can help to improve the genetic resources of the species and understand the reasons for the trait alternations between different varieties. Therefore, we also sequenced and de novo assembled genome of a L. japonica variety that is called ‘Sijihua’, which is largely planted in Pingyi county, Shandong province. It’s reported that Sijihua are the most cold-resistant variety by studying different varieties of L. japonica in different regions10.
Here, we generated a chromosome-scale of the genome assembly of the variety of Sijihua using the combination of PacBio long reads, Illumina reads and the Hi-C sequencing data. Approximately 886.04 Mb genome was assembled with the contig N50 length of 1.58 Mb. A total of 826.50 Mb (93.28%) of the assembled sequences were anchored to 9 pseudo-chromosomes (Table 1). We predicted 39,320 protein-coding genes, and 92.87% of each gene were assigned by BLASTP against NR, GO, KOG, KEGG and other databases. We identified 644 tRNAs, 2,156 rRNAs, 109 miRNAs and 5,502 pseudogenes (Table 2). We also identified 255,264 simple sequence repeats (SSRs), and 40,252 are polymorphic SSRs (Table 3). The genome assembly of variety Sijihua is a valuable material to the germplasm resources of L. japonica, and helps researchers to explore the specificity of different varieties. The results also provide valuable clues to the molecular basis of cold-resistance traits of Sijihua and will facilitate further genetic improvements.
Methods
Sample collection, library construction and genome size estimation
High-quality genomic DNA was extracted from young-fresh leaf tissue of Sijihua using CTAB (cetyl trimethylammonium bromide) method, and the samples were collected from the Zhongke Honeysuckle Planting Cooperative in Pingyi County, Shandong, China (Fig. 1). The qualified genomic DNA was broken to the target fragment (350 bp) by ultrasonic shock and the Illumina library was constructed through end repairing, adding 3’ A tail, ligating adapters and enriching with PCR. The fragment size and quality of the library were detected by 2100 and Q-PCR. Next, the sequencing of the library were performed by using Illumina Novaseq 6000, which finally generated 31.48 million reads, 50.26 Gb of raw data, which covered 61.48 × of the genome. PacBio library were constructed by using BulePippin to screen the target fragment that was interrupted by g-tube, subsequently was sequenced with PacBio Sequel II system. Consequently, the two SMRT-cells generated a total of 4,461,375 reads with N50 size of 29,096 bp. Totally we got 87.61 Gb sequencing data, accounting for 98.88 × of the entire genome. Fresh leaf tissue of honeysuckle was used to construct a library for Hi-C analysis. The fresh tissue was fixed with formaldehyde, attaining interacting loci to be bound to one another, and then cross-linked DNA was digested by restriction enzyme Hind III. Sticky ends were labeled with biotin during its repairing. Next, the interacting DNA fragments were ligated, purified and finally broke them into 300 bp ~ 700 bp fragments. Each ligated DNA fragment was marked with biotin and streptavidin beads were used to pull-down the interacting DNA fragments to complete the Hi-C library construction. The libraries were then sequenced on Illumina Novaseq 6000 platform, which generated 307,357,239 pairs of reads and 91.84 Gb clean data, which cover 103.65x of the genome.

The five growth stages of honeysuckle. (a) The juvenile bud stage. (b) The third green stage. (c) The complete white stage. (d) The silver flowering stage. (e) The gold flowering stage.
A k-mer (k = 19) analysis was constructed using 61.48 × Illumina data to estimate the genome size, proportion of repeat sequence and heterozygosity11. From the 19-kmers distribution, we could estimate the heterozygosity and repeat ratio of the Sijihua genome to be 0.74% and 51.8%, respectively and the estimated genome size was 817.45 Mb (Fig. 2 and Table 1).

19-kmer distribution in the honeysuckle genome.
RNA sequencing and analysis
Total RNA was extracted by using RNeasy Plus Mini Kit (Qiagen) from different development stages, including juvenile bud (JB), green bud (GB), white bud (WB), silver flower (SF), and golden flower (GF). Libraries were constructed by TruSeq RNA Library Prep Kit v.2 (Illumina, San Diego, CA, USA). Each library was constructed with three biological replicates, and 150 bp paired-end reads were sequenced by using the Illumina NovaSeq 6000 platform. Raw reads were trimmed by using TRIMMOMATIC (v.0.39)12, and the clean reads were aligned to the reference genome by HISAT2 (v. 2.2.1)13 with default parameters, and only uniquely mapped reads were kept. Expression value was estimated by StringTie (v. 2.1.5)14 as FPKM (Fragments per kilobase of exon model per million reads mapped). Genes with FPKM > 0.5 were considered as expressed, and used for further analysis. Differentially expressed genes (DEGs) were identified by DESeq2 (v 1.28.1)15 with default parameters. RNA-seq data of LJ10107428 were downloaded from National Genomics Data Center (https://ngdc.cncb.ac.cn) with BioProject ID (PRJCA001719).
De novo genome assembly
PacBio long reads were error corrected using Canu v1.516 and the top 40 x coverage of the longest corrected reads were subsequently assembled by SMARTdenovo17. To improve the accuracy of the assembly, the 61.48x Illumina short reads used in the genome survey were applied to three rounds of correction by Pilon v1.2218. The high-quality Hi-C reads were used to cluster, order and orient the contigs onto pseudo-chromosomes by using the LACHESIS19. We preliminary assembled the PacBio long reads into contig sequences of 886.04 Mb, including 1,159 contigs with N50 of 1.58 Mb, and the longest contig is 12.45 Mb. These contigs were further anchored onto 9 pseudo-chromosomes, accounting for 93.28% of the assembled genome. The final chromosome-scale genome assembly of Sijihua was 886.13 Mb with a scaffold N50 of 79.57 Mb (Table 1).
Repeat annotation
De novo and structure-based predictions were integrated to annotate repetitive sequences. LTR_FINDER v1.0520 and RepeatScout v1.0521 were primarily used to build a de novo repeat sequences library of Sijihua genome, which was classified by using PASTEClassifier v1.022 and merged with Repbase v19.0623 database as the final repeat sequences database. Structure-based predictions were performed by using RepeatMasker v4.0524 based on the constructed repeat sequences database. We identified 573.89 Mb (64.76%) of repetitive sequences in Sijihua genome. Most of these repeat sequences are Class I (53.57%) retrotransposons, including Copia, Gypsy, LINE and SINE, accounted for 15.93%, 19.14%, 2.61% and 0.34% of the entire genome, respectively. In addition, Class II DNA transposons make up 5.82% of the genome (Table 2).
Protein-coding genes prediction and other annotations of the genome
Prediction of protein-coding genes was based on ab initio gene predictions, homology-based predictions and transcriptome-based predictions. Ab initio predictions were performed by Genscan v3.125, Augustus v3.126, GlimmerHMM v1.227 v3.0.4, GeneID28 v1.4, and SNAP (version 2006–07–28)29. For homology-based prediction, GeMoMa v1.3.130 was used to annotate the gene models in Sijihua using amino acid sequences from Daucus carota, Helianthus annuus, Lactuca sativa, Lonicera japonica and Arabidopsis thaliana genome. For RNA-seq-based prediction, the clean RNA-seq reads were aligned to the assembled genome using Hisat31 v2.0.4 and Stringtie32 v1.2.3, and then TransDecoder33 v2.0, GenemarkS-T34 v5.1 and PASA35 v2.0.2 were jointly used for final coding-gene prediction. Finally, 39,320 gene models were predicted after integrating results of the three methods of predictions by EVidenceModeler36 v1.1.1 (Table 2). For non-coding RNAs annotation, microRNA and rRNA were detected by aligning the assembled genome against the to Rfam37 database using BLASTN. tRNA was identified by tRNAscan-SE38. Finally, we totally identified 2,909 non-coding RNAs, including 109 miRNAs, 2,156 rRNAs and 644 tRNAs.
The sequence of pseudogenes is similar to that of functional genes, but whose original function is lost due to insertions, deletions and other variants. The predicted protein sequences were used to search for homologous gene sequences on the genome through BLAT39 alignment, and then GeneWise39 was used to search for immature stop codons and frameshift mutations in gene sequences. In total, 5,502 pseudogenes were predicted (Table 2).
For gene functional annotation, we aligned the predicted protein-coding gene sequences against public functional databases using BLAST40 v2.2.31 (-evalue 1E-5), such as NR41, KOG42, GO43, KEGG44 and TrEMBL45. In addition, the motif and domain information were annotated using InterProScan46 through searching against public databases, including PROSITE47, HAMAP48, Pfam49, PRINTS50, ProDom51, SMART52, TIGRFAMs53, PIRSF54, SUPERFAMILY55, CATH-Gene3D56 and PANTHER57. As a result, more than 92% of protein-coding genes were annotated, and 1,376 conserved motifs and 36,282 domains were identified.
Identification of SSRs and polymorphism
Reference genomes of Sijihua and Lj10107428 were analyzed for the identification of simple sequence repeats (SSRs) and polymorphism by using CandiSSR58 with default parameters. We identified 255,264 and 182,207 SSRs in Sijihua and Lj10107428 genomes, respectively. Moreover, we found 40,252 SSRs are polymorphic between Sijihua and Lj10107428 genomes (Table 3). In addition, among all the SSRs identified in Sijihua genome, the most abundant SSRs motifis are di-nucleotide (192,362), followed by tri-nucleotide (54,009), tetra-nucleotide (6,395), penta-nucleotide (1,526), and hexa-nucleotide (972).
Global genome comparison of the Lj10107428 and Sijihua
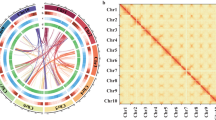
Genome comparison between Sijihua and Lj10107428 was performed by using the NUCmer program embedded in MUMmer4 with parameters –mum –l 40 –c 100, then the delta alignment file was filtered by delta-filter with parameters -1, and finally show-snps was used to identify single nucleotide polymorphisms (SNPs) with parameters -ClrT. SyRI v1.559 was used to extract structure variations (SVs) based on the alignment file with tab-delimited text format performed by show-coords program. MCScanX60 was used to identify gene collinearity, including within and between genomes. In total, we identified 2,996,015 SNPs between Sijihua and Lj10107428. Among them, 14.44% of the SNPs were located in the genic region. Comparison between Lj10107428 and Sijihua genomes, we found 5,150 small insertions/deletions (indels, length shorter than 500 bp), and more than 4.5 Mb of presence/absence variation (PAV, length longer than 500 bp). Notably, we identified 895 Sijihua-specific genomic sequences (2.61 Mb in total), and 418 Lj10107428-specific genomic sequence (1.84 Mb in total) longer than 500 bp. These PAV segments were unevenly distributed across the chromosomes, and the longest PAV sequence was a 261 Kb segment on chromosome 2 (Fig. 3a). In addition, we also found 156 inversions ( >1,000 bp) and several translocations ( >10 kb) between these two genomes. The genome sequence comparison between Lj10107428 and Sijihua reveals high collinearity. Although some structure variations were detected between these two genomes, they primarily consist of large syntenic block with high degrees of collinearity (Fig. 3b). In addition, we identified 301 large syntenic blocks between Sijihua and Lj10107428, containing 25,128 syntenic genes. Specificity, there is an inverted region of approximate 19 Mb on chromosome 8 between Sijihua and Lj10107428 genomes (Fig. 3b). We also noticed that 561 and 302 NBS-LRR genes located in to Sijihua and Lj10107428 genomes, respectively (Fig. 3b). By comparing the annotated protein-coding genes, we found that 26,937 genes of Sijihua and Lj10107428 were shared by reciprocal best hit of BLAST algorithm with parameter (E-value < 1e-10). More species-specific genes were found in Sijihua (12,383) than that in Lj10107428 (6,361) (Fig. 3c).

Comparative genomic analysis between Sijihua and Lj10107428 varieties of honeysuckle. (a) Genomic features landscape of the Sijihua genome. Density of genes, TEs, SNPs, indels, PAVs, inversions and translocations were calculated in a 500 Kb sliding window. (b) Gene collinearity between Sijihua and Lj10107428 varieties. NBS-LRR genes were annotated as yellow dot across genome. (c) Venn diagram of the overlapped genes between Sijihua and Lj10107428 genomes. (d) Expression level comparison of shared genes in Sijihua and Lj10107428 varieties.
From the 18,958 overlapped expressed (FPKM > 0.5) genes between Sijihua and Lj10107428, most of them (>93.4%) did not show differential expression variation at juvenile bud stage. However, hundreds of genes were differential expressed, including 16 NBS-LRR genes. Notably, 11 of 16 NBS-LRR genes were highly expressed in Sijihua at juvenile bud stage. This observation is consistent across other flower development stages (Fig. 3d).
Data Records
The raw data of PacBio, Illumina and Hi-C sequencing were submitted to the National Center for Biotechnology Information (NCBI) SRA database with accession number SRP35369861 under BioProject accession number PRJNA794868. RNA-seq data were deposited into the NCBI (accession number PRJNA813701)62. The assembled genome had been deposited at GenBank with accession number SAMN2466218463. In addition, the genome annotation file had been submitted at the Figshare64.
Technical Validation
Evaluation of the genome assembly
To evaluate the quality of genome assembly, bwa (version: 0.7.10-R789; mode: aln) was used to align the Illumine short reads with the reference genome, and 99.75% of the Illumina short reads were mapped to the reference genome CEGMA65 v2.5 was used to assess the integrity of the final genome assembly. The CEGMA database contained 458 conserved core eukaryotic genes, while our assembled genome contained 439 (95.85%), which suggested that our assembled genome contains most of the core eukaryotic genes. BUSCO66 v4.0 was used to assess the integrity of our genome assembly by using the Embryophyta database of OrthoDB v10. Of the 1614 expected embryophyta genes, our genome contains 1,556 (97.03%). Together, these three evaluation systems demonstrate the high integrity of our assembled genome (Table 1).
Furthermore, to assess the result of Hi-C assembly, and the number of Hi-C read pairs coverage between any two bins acts as a strength signal of interaction between the two bins. As chromosomal interaction heatmap shown, within each group, it was found that the intensity of interaction at the diagonal position was higher than that at the non-diagonal position (Fig. 4), which was consistent with the principle of Hi-C-assisted genome assembly and proved that the genome assembly was accurate.

Hi-C contact map of the chromosome-scale assembly of Sijihua. Hi-C interaction matrix shows the pairwise correlations among 9 pseudomolecules. The intensity of the dark color is scaled to the strength of the correlation.
Code availability
BUSCO: --evalue 1e-03, -sp Arabidopsis.LACHESIS:CLUSTER_MIN_RE_SITES = 30;CLUSTER_MAX_LINK_DENSITY = 2;CLUSTER_NONINFORMATIVE_RATIO = 2;ORDER_MIN_N_RES_IN_TRUN = 49;ORDER_MIN_N_RES_IN_SHREDS = 49.Software parameters of repeat annotation: default parameters for LTR_FINDER, RepeatScout, and PASTEClassifier. RepeatMasker: -nolow -no_is -norna -engine wublast.Software parameters of gene prediction: default parameters for Genscan, Augustus,GlimmerHMM, GeneID, SNAP, GeMoMa, Stringtie, TransDecoder, GeneMarkS-T, and EVM.Hisat:--max-Intronlen 20000, --min-intronlen 20. PASA: -align_tools gmap, -maxintronlen20000.GenBlastA: -e 1e-5.BLASTP: -e 1e-10.CandiSSR: perl CandiSSR.pl -i crl.file -o out_path/.Default parameters were used in other software unless otherwise specified.
References
Shang, X., Pan, H., Li, M., Miao, X. & Ding, H. Lonicera japonica thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. Journal of Ethnopharmacology 138 (2011).
Schierenbeck, K. A. Japanese honeysuckle (Lonicera japonica) as an invasive species; history, ecology, and context. Critical Reviews in Plant Sciences 23 (2010).
Leatherman, A. D. Ecological life-history of Lonicera japonica thunb. (1955).
Wang, Y. Research on Chinese medicine honeysuckle medicinal ingredients and pharmacological effects. Adv Eng Res 122, 42–45 (2017).
Machida, K., Sasaki, H., Iijima, T. & Kikuchi, M. Studies on the constituents of Lonicera species. XVII. New iridoid glycosides of the stems and leaves of Lonicera japonica thunb. Chemical and pharmaceutical bulletin 50, 1041–1044 (2002).
Rai, A. et al. De novo transcriptome assembly and characterization of Lithospermum officinale to discover putative genes involved in specialized metabolites biosynthesis. Planta medica 84, 920–934 (2018).
Zhang, L. et al. Different gene expression patterns between leaves and flowers in Lonicera japonica revealed by transcriptome analysis. Frontiers in plant science 7, 637 (2016).
He, L. et al. Transcriptome analysis of buds and leaves using 454 pyrosequencing to discover genes associated with the biosynthesis of active ingredients in Lonicera japonica thunb. PLoS One 8, e62922 (2013).
Pu, X. et al. The honeysuckle genome provides insight into the molecular mechanism of carotenoid metabolism underlying dynamic flower coloration. New Phytologist 227, 930–943 (2020).
Wang, H. Analysis on cold resistance of honeysuckle from different habitats. Northern Horticulture, 139-141 (2014).
Xiao, Y., Xiao, Z., Ma, D., Liu, J. & Li, J. Genome sequence of the barred knifejaw oplegnathus fasciatus (Temminck & Schlegel, 1844): The first chromosome-level draft genome in the family Oplegnathidae. GigaScience 8, giz013 (2019).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research 27, 722–736 (2017).
Ruan, J. SMARTdenovo: Ultra-fast de novo assembler using long noisy reads. Github Available at: https://github.com/ruanjue/smartdenovo[Accessed January 10, 2019] (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963 (2014).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology 31, 1119–1125 (2013).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PloS one 9, e91929 (2014).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology 268, 78–94 (1997).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Blanco, E., Parra, G. & Guigó, R. Using geneid to identify genes. Current protocols in bioinformatics 18, 4.3. 1–4.3. 28 (2007).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 1–9 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic acids research 44, e89–e89 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295 (2015).
Haas, B. & Papanicolaou, A. TransDecoder (find coding regions within transcripts) http://transdecoder.github.io.
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic acids research 43, e78–e78 (2015).
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC genomics 7, 1–17 (2006).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic acids research 33, D121–D124 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964 (1997).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome research 12, 656–664 (2002).
Altschul, S., Gish, W., Miller, W., Myers, E. & Lipman, D. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Marchler-Bauer, A. et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic acids research 39, D225–D229 (2010).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome biology 5, 1–28 (2004).
Dimmer, E. C. et al. The UniProt-GO annotation database in 2011. Nucleic acids research 40, D565–D570 (2012).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370 (2003).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Bairoch, A. PROSITE: a dictionary of sites and patterns in proteins. Nucleic acids research 19, 2241 (1991).
Lima, T. et al. HAMAP: a database of completely sequenced microbial proteome sets and manually curated microbial protein families in UniProtKB/Swiss-Prot. Nucleic acids research 37, D471–D478 (2009).
Finn, R. D. et al. Pfam: clans, web tools and services. Nucleic acids research 34, D247–D251 (2006).
Attwood, T. & Beck, M. PRINTS–a protein motif fingerprint database. Protein Engineering, Design and Selection 7, 841–848 (1994).
Bru, C. et al. The ProDom database of protein domain families: more emphasis on 3D. Nucleic acids research 33, D212–D215 (2005).
Letunic, I. et al. SMART 4.0: towards genomic data integration. Nucleic acids research 32, D142–D144 (2004).
Haft, D. H., Selengut, J. D. & White, O. The TIGRFAMs database of protein families. Nucleic acids research 31, 371–373 (2003).
Wu, C. H. et al. PIRSF: family classification system at the protein information resource. Nucleic acids research 32, D112–D114 (2004).
Gough, J. & Chothia, C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic acids research 30, 268–272 (2002).
Lees, J. et al. Gene3D: a domain-based resource for comparative genomics, functional annotation and protein network analysis. Nucleic acids research 40, D465–D471 (2012).
Thomas, P. D. et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic acids research 31, 334–341 (2003).
Xia, E. H. et al. CandiSSR: An Efficient Pipeline used for Identifying Candidate Polymorphic SSRs Based on Multiple Assembled Sequences. Front Plant Sci 6, 1171, https://doi.org/10.3389/fpls.2015.01171 (2015).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome biology 20, 1–13 (2019).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49–e49 (2012).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP353698 (2022).
NCBI BioProject https://identifiers.org/bioproject:PRJNA813701 (2022).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_021464415.1 (2022).
Lai, K. Genome annotation for Sijihua, which is a stress-resistance honeysuckle variety. figshare https://doi.org/10.6084/m9.figshare.18092708.v6. (2022).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32160142 to H.W. and No.81873095 to K.G.) and Science and Technology Major Project of Guangxi (Gui ke 2018–266-Z01) to H.W. H.Y. is supported by Innovation Project of Guangxi Graduate Education (YCBZ2021005).
Author information
Authors and Affiliations
Contributions
H.W. and N.C. conceived the study and supervised all parts of the project. K.G., N.C. and Z.X. collected the samples, extracted the genomic DNA and conducted sequencing. H.Y. performed comparative transcriptome analysis. H.Y., K.L. and K.G. performed comparative genomic analysis. K.L., H.W. and M.A.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, H., Guo, K., Lai, K. et al. Chromosome-scale genome assembly of an important medicinal plant honeysuckle. Sci Data 9, 226 (2022). https://doi.org/10.1038/s41597-022-01385-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01385-4
