
Abstract
Genes are pleiotropic and getting a better knowledge of their function requires a comprehensive characterization of their mutants. Here, we generated multi-level data combining phenomic, proteomic and metabolomic acquisitions from plasma and liver tissues of two C57BL/6 N mouse models lacking the Lat (linker for activation of T cells) and the Mx2 (MX dynamin-like GTPase 2) genes, respectively. Our dataset consists of 9 assays (1 preclinical, 2 proteomics and 6 metabolomics) generated with a fully non-targeted and standardized approach. The data and processing code are publicly available in the ProMetIS R package to ensure accessibility, interoperability, and reusability. The dataset thus provides unique molecular information about the physiological role of the Lat and Mx2 genes. Furthermore, the protocols described herein can be easily extended to a larger number of individuals and tissues. Finally, this resource will be of great interest to develop new bioinformatic and biostatistic methods for multi-omics data integration.
Measurement(s) | preclinical phenotype • protein expression profiling • metabolite profiling |
Technology Type(s) | phenotyping tests • mass spectrometry assay |
Factor Type(s) | genotype • sex |
Sample Characteristic - Organism | Mus musculus |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.15015273
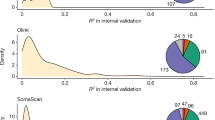
Similar content being viewed by others


Background & Summary
The large scale analysis of gene function ongoing in the International Mouse Phenotyping consortium (www.mousephenotype.org) has consolidated the pleiotropic hypothesis of gene function in mammals1,2,3. Thus, more comprehensive approaches are needed to investigate gene’s function. The global study of proteins4 and metabolites5,6 are two major approaches for the understanding of biological processes and metabolism. Moreover, metabolites interact closely with proteins in the cell, either as substrates, cofactors and enzyme products, or as allosteric regulators of enzymes, transmembrane receptors or transcription factors7. Joint proteomic and metabolomic characterization therefore represents a unique opportunity to bring new information on the physiological processes under the control of genes on an integrated scale. The potential of this combined approach has been shown recently, with the identification of markers for hepatic lipotoxicity by the global analysis of proteins and lipids in 107 murine lines8, as well as the demonstration of significant variations in proteins and metabolites from the same immune pathways in severe forms of COVID-199.
Furthermore, the availability of combined proteomic and metabolomic data is of interest to define multi-omics signatures that may predict more efficiently disease development and progression10. As an example, the statistical integration of proteomic, metabolomic and lipidomic data (by concatenating selected variables using a Naïve Bayes classifier, or by fusing Random Forest and Linear Discriminant Analysis models learned from each of the data blocks) has proven to provide a better predictive performance than that obtained with each of the individual type of blocks in the case of type 1 diabetes status11.
The interest in integrating proteomic and metabolomic data also stems from the common technology used in both approaches, i.e. liquid chromatography coupled to high-resolution mass spectrometry12. At the computational level, common formats exist for raw and pre-processed data, such as mzML13 and mzTab14, respectively. Computational tools which can process either types of mass spectrometry raw data are already available15,16,17,18. Moreover, sample preparation protocols for the simultaneous extraction of proteins and metabolites have been proposed19,20,21 enabling to combine both omics within an unique analytical strategy.
While a few studies have reported the relevance of integrating proteomic and metabolomic approaches for obtaining deeper insight into disease development or into the underlying biochemical mechanisms, the routine use of such a combined strategy is not straightforward for the deep phenotyping of large cohorts. Here we address the possibility of generating and making publicly available a dataset combining both approaches in a fully non-targeted, standardized, and reproducible way.
As part of the large-scale characterization of mouse models in which each gene is inactivated3,22, we focused on two knock-out mouse models for the Lat (linker for activation of T cells; MGI:1342293) and Mx2 genes (MX dynamin-like GTPase 2; MGI:97244), respectively, as well as the control line (WT), generated by the Phenomin-ICS infrastructure, member of the International Mouse Phenotyping Consortium (IMPC). On the one hand, Lat, besides its role in T-cell receptor (TCR) signalling23, has been shown to be involved in neurodevelopmental diseases24. On the other hand, Mx2 is one of the coding genes in the genome region modelling Down syndrome in mice25. The characterization of mouse lines is currently based on a battery of animal phenotypic tests (anatomy, behaviour, histology, haematology, physiology), the results of which feed the IMPC database (https://www.mousephenotype.org). To further characterize these models, global molecular approaches are required26. The originality of our study is to provide, in addition to the preclinical data, a comprehensive molecular characterization by proteomic and metabolomic analyses of liver and plasma samples from the Lat, Mx2 and WT mouse models.
Our study consists of 9 datasets (1 preclinical, 2 proteomics and 6 metabolomics), generated by the four French infrastructures for mouse phenogenomics, proteomics, metabolomics and bioinformatics, and are publicly available in the ProMetIS R package27 to ensure accessibility, interoperability, and reusability following the FAIR principles28. The dataset provides access to unique molecular functional information on the Lat and Mx2 genes. Furthermore, the protocols and computational workflows provided here can be considered as generics, and as such, they can be easily extended to a larger number of individuals and tissues. In particular, this pilot study paves the way for the inclusion of proteomics and metabolomics analyses in the standardized IMPC pipelines for the characterization of mouse mutants. Finally, the ProMetIS resource will be of great interest to develop new bioinformatic and biostatistic methods for the processing and integration of (pre)clinical, proteomic and metabolomic approaches.
Methods
Preclinical data
Mouse lines
The Latem1(IMPC)Ics and Mx2em1(IMPC)Ics homozygote mouse mutant lines were generated at the Mouse Clinical Institute in Illkirch, France (Phenomin-ICS, http://www.ics-mci.fr), as part of the International Mouse Phenotyping Consortium (IMPC)29. Briefly, mice were generated on a pure C57BL/6 N background using CrispR/Cas9. gRNAs were selected with the Crispor program (http://crispor.tefor.net/crispor.py) to delete a critical exon, here exon 2 for both Lat and Mx2 genes, that will introduce a change in the open reading frame and the stop of translation. After microinjection in the pronucleus of C57BL/6 N fertilized eggs, 16 pups were born in the Lat study, and 6 of them had the expected deletion of exon 2 of Lat. A line was established from one founder (#13), the size of the deletion was 242 bps. Primers F1 (CTTCTTGGTCACGCTCCTGGCTG) and R1 (ATGCTTCTTGGGTACAAACTGGCAG) were used for genotyping (WT allele: 600 bps, KO allele: 358 bps). For the Mx2 gene, 15 pups were born after microinjection in C57BL/6 N fertilized eggs and 2 carried the expected deletion of exon 2. A line was established from one founder (#9), the size of the deletion was 411 bps. Primers F1 (TGGAACAGACACCTAAGTCTTGGTC) and R6 (CAGACACCAAGTGGCTTCTCCCAGG) were used for genotyping (WT allele: 856 bps, KO allele: 445 bps). The two mouse mutant mice were bred to homozygosity in 2 generations on the C57BL/6 N genetic background and maintained in a temperature-controlled facility (20–22 °C) on a 12‐hour light/dark cycle (7AM-7PM) with free access to standard chow diet (D04, SAFE Villemoisson-sur-Orge, France). The homozygous animals, 6 to 8 males and females, from the Lat and Mx2 KO lines, were obtained from homozygotes breeding at the 3rd generation. Wild-type control individuals were derived from the C57BL/6 N colony maintained under the same conditions (Fig. 1).

Experimental design. A total of 42 mice from the Lat−/− and Mx2−/− genotypes (as well as the wild-type controls), and from both sex, were analyzed by a series of phenomic (preclinical) measurements. Liver and plasma samples from all mice were further analyzed at the molecular level by proteomics and metabolomics, except for 6 plasma samples which could be not analyzed by proteomics due to the limited volume available (Lat: 1 male and 1 female, Mx2: 1 male and 1 female, WT: 2 males).
Sample collection
At the end of the phenotyping pipeline (16 weeks of age), blood was collected by retro orbital puncture under isoflurane anaesthesia for biochemistry and haematology analysis according to the “Clinical Chemistry” protocol available on the IMPReSS database (see the Supplementary File 3). Briefly, 160–200 μl of plasma were collected in a gel tube containing lithium Heparin in the morning. Whole blood samples were centrifuged for 10 minutes at 5,000 g in a refrigerated centrifuge set at 8 °C. The plasma samples were aliquoted and frozen (−20 °C) until transfer to the partners for metabolomic and proteomic analyses. Due to limited volume, 6 plasma samples (Lat: 1 male and 1 female, Mx2: 1 male and 1 female, WT: 2 males) were not analyzed by proteomics. Upon sacrifice, i) all animals were weighted and measured, ii) an extensive necropsy was performed on two mice per sex and genotype, and iii) all organs defined in IMPC protocol were collected on these animals. The liver tissues from all animals were dissected, snap-frozen in liquid N2, and transferred to partners for metabolomics and proteomics analysis.
Phenotyping
Phenotyping data were collected between the age of 4 and 16 weeks (Fig. 2). Both mutant and wild-type mice were tested through a broad-based primary phenotyping pipeline in all the major adult organ systems and most areas of major human diseases. Phenotyping tests are standardized and cross-validated between centres of the consortium30, and all procedures used to generate data from mutant and wild-type control mice followed the defined and validated Phenomin-ICS protocols available on the IMPReSS database (https://www.mousephenotype.org/impress; see the Supplementary File 3 for the list of identifiers and links to the specific procedures).

The 200 phenotypic (preclinical) measurements cover a large panel of anatomical, behavioural, histological, haematological, and physiological tests (see the Supplementary File 3 for the identifiers and the links to the experimental procedures in the online IMPReSS database).
Ethical statement
Phenomin-ICS is accredited by the French Ministry of Higher Education, Research and Innovation, and the French Ministry of Agriculture (agreement #A67-218-37), and in accordance with the Directive of the European Parliament: 2010/63/EU, revising/replacing Directive 86/609/EEC and with French Law (Decree n°2013-118 01 and its supporting annexes entered into legislation 01 February 2013) relative with the protection of animals used in scientific experimentation. All animal experiments were approved by local ethical committees: Approval Committee: Com’Eth N°17 and French Ministry of Higher Education, Research and Innovation (Approval license: MESR: APAFIS#4789-2016040511578546). Animal studies were supervised in compliance with the European Community guidelines for laboratory animal care and use. Every effort was made to minimize the number of animals used and their suffering.
Proteomics
Sample preparation
Mice liver proteins were extracted in 10% w/v Laemmli buffer (Tris 10 mM pH 6.8, EDTA 1 mM, beta-mercaptoethanol 5%, SDS 5%, glycerol 10%) with 1% v/v protease inhibitors (Pierce #P8340), firstly under gentle agitation at room temperature for 2 hours, and then using quick sonication on ice (3 × 10 s, 135 W) to ensure complete solubilisation. Samples were centrifuged at 10,000 g for 10 min to remove possible cell debris. Protein concentrations in the supernatants were determined using the RC-DC Protein Assay (BioRad, Hercules, CA, USA). Then 1% blue bromophenol was added, the samples were heated at 95 °C for 5 min, 50 µg of sample were loaded on an in-house prepared 4% acrylamide SDS-PAGE stacking gel and run at 50 V for 13 min. Proteins were fixed and then stained using colloidal Coomassie blue (Fluka, Buchs, Switzerland) for 5 min. The stacking bands were manually excised, destained, reduced for 1 hour with 10 mM dithiothreitol, and alkylated for 20 min with 55 mM iodoacetamide in the dark. Overnight digestion was then performed at 37 °C using 1:50 enzyme:protein ratio of Trypsin/Lys-C Mix (#V507A, Promega, Madison, USA). Peptides were extracted during 1 hour with 160 µl acetonitrile (ACN). Organic solvent was eliminated using a vacuum centrifuge (SpeedVac, Savant, Thermo scientific, Waltham, MA, USA), and peptides were re-suspended in 140 µl of water acidified with 0.1% formic acid (FA).
For analysis of plasma proteins, filter-aided sample preparation was performed using Amicon Ultra devices (0.5 mL, cutoff 10 kDa, Merck Millipore). Three microliters of each plasma sample were diluted with 350 µl of 4 M urea, 50 mM ammonium bicarbonate buffer and centrifuged in the device at 14,000 g and 4 °C, to a concentrated volume of 50 µl. For reduction of cysteine residues, 350 µl of 10 mM TCEP in 4 M urea, 50 mM ammonium bicarbonate buffer were added on the device, and the samples were incubated 10 min at room temperature. Sample volume was reduced again down to 50 µl by centrifugation at 14,000 g and 4 °C. Alkylation of cysteine residues was performed by addition of 350 µl of 55 mM iodoacetamide in 4 M urea, 50 mM ammonium bicarbonate buffer, followed by 15 min incubation in the dark, and concentration of the samples by centrifugation of the filter device. Two additional washes of the samples were performed with 4 M urea, 50 mM ammonium bicarbonate buffer. Proteins were digested with 1:30 enzyme:protein ratio of Trypsin/Lys-C Mix (Promega) in 4 M urea, 50 mM ammonium bicarbonate, for 2 h at 37 °C. The concentration of urea was then reduced to 1 M by dilution with 50 mM ammonium bicarbonate, and the samples were further incubated for 3 h at 37 °C. Resulting peptides were collected by centrifugation of the 10 kDa filter, which was further rinsed with 100 µl of 0.5 M NaCl. Peptides were desalted on C18 spin columns (Pierce), dried in a vacuum centrifuge, and re-suspended in 180 µl of water containing 2% ACN and 0.1% trifluoroacetic acid (TFA).
Before LC-MS analysis, a set of reference peptides (iRT kit; Biognosys AG, Schlieren, Switzerland) was added to all samples. Furthermore, for the quality control of the mass spectrometry analysis sequence, a pooled quality control (QC) sample was constituted by pooling 1 µl of each sample, either for the liver tissue samples or for the plasma samples series. The samples were stored at 4 °C and analyzed within a week.
Analytical chemistry
NanoLC-MS/MS analyses of the tryptic peptides obtained from liver proteins were performed on a nano-UPLC system (nanoAcquityUPLC, Waters, USA) coupled to a quadrupole-Orbitrap hybrid mass spectrometer (Q-Exactive plus, Thermo Scientific, San Jose, CA). Briefly, 1 µl (320 ng) of each sample was concentrated/desalted on a trap column (Symmetry C18, 180 μm × 20 mm, 5 µm; Waters) using 99% of solvent A (0.1% FA in water)/1% solvent B (0.1% FA in ACN) at a flow rate of 5 µl/min for 3 minutes. Afterwards, peptides were eluted from the separation column (BEH130 C18, 75 μm × 250 mm, 1.7 μm; Waters) maintained at 60 °C using a 79 min gradient from 1–35% of B at a flow rate of 450 nL/min. The Q-Exactive Plus was operated in positive ion mode with source temperature set to 250 °C and spray voltage to 1.8 kV. The mass spectrometer was operated in data-dependent acquisition mode, and spectra were acquired through automatic switching between full MS and MS/MS scans. Full scan MS spectra (300–1800 m/z) were acquired at a resolution of 70,000 at m/z 200 with an automatic gain control (AGC) value set to 3 × 106 ions, a maximum injection time set to 50 ms, and the lock-mass option enabled (polysiloxane, 445.12002 m/z). Up to 10 most intense multi-charged precursors per full MS scan were isolated using a 2 m/z window and fragmented using higher energy collisional dissociation (HCD, normalized collision energy of 27 eV). MS/MS spectra were acquired at a resolution of 17,000 at m/z 200 with an AGC value set to 1 × 105, a maximum injection time set to 100 ms, and the peptide match selection option was turned on. Dynamic exclusion of already fragmented precursors was set to 60 seconds. The system was fully controlled by the Xcalibur software (v3.1.66.10; Thermo Fisher Scientific). MS data were saved in .raw file format (Thermo Fisher Scientific) using XCalibur.
For analysis of plasma samples, peptides were separated using a nano-UPLC system (Ultimate NCS-3500RS System; Thermo Fisher Scientific) coupled to a quadrupole-Orbitrap hybrid mass spectrometer (Q-Exactive HFX, Thermo Scientific, San Jose, CA). Five microliters of each sample were loaded on a C18 precolumn (300 µm inner diameter × 5 mm, Thermo Fisher Scientific) in a solvent made of 2% ACN and 0.05% TFA, at a flow rate of 20 µl/min. After 3 min of desalting, the precolumn was switched online with the analytical C18 column (75 µm inner diameter × 50 cm, Acclaim PepMap C18, 2 µM, Thermo Fisher Scientific), equilibrated in 90% solvent A (5% ACN, 0.2% FA) and 10% solvent B (80% ACN, 0.2% FA), in order to speed-up the elution of the peptides at the beginning of the analytical run. Peptides were then eluted by a gradient composed of 2 slopes (from 8 to 24% of ACN during 50 min, and from 24% to 36% of ACN during 10 min), at a flow rate of 350 nl/min. Full scan MS spectra (350–1400 m/z) were acquired with a resolution of 60,000 and an AGC target of 3 × 106 ions. The 6 most intense ions were selected (1.3 m/z window) for fragmentation by high energy collision induced dissociation (normalized collision energy of 28 eV), and the resulting fragments were analysed at a resolution of 30000, using an AGC target of 1 × 105 and a maximum fill time of 54 ms. Dynamic exclusion was used within 30 s to prevent repetitive selection of the same peptide.
All sample analyses, for either the liver or plasma series, were randomly ordered (considering the genotype, sex and weight of the mice). Pooled QCs were injected every 5 samples. To minimize carry-over, one solvent blank injection was included between all samples.
Data processing
Spectrum identification: Raw files were converted to.mgf peaklists using MsConvert and were submitted to Mascot database searches (version 2.5.1, MatrixScience, London, UK) against a murine protein sequences database downloaded from the SwissProt website (2018_08_20), to which common contaminants, iRT and decoy sequences were added. The concatenated database contains 2 × 17 111 protein entries. Spectra were searched with a mass tolerance of 5 ppm in MS mode and 0.07 Da in MS/MS mode. Two trypsin missed cleavages were tolerated. Carbamidomethylation of cysteine residues was set as a fixed modification. Oxidation of methionine residues and acetylation of proteins n-termini were set as variable modifications. Identification results were imported into the Proline software (http://proline.profiproteomics.fr)31 for validation. Peptide Spectrum Matches (PSM) with pretty rank equal to one were retained. False Discovery Rate was then optimized to be below 1% at PSM level using Mascot Adjusted E-value and below 1% at Protein Level using Mascot Mudpit score. Label Free Quantification: Peptides Abundances were extracted thanks to the Proline software31 version 1.6 using an m/z tolerance of 5 ppm. Alignment of the LC-MS runs was performed using Loess smoothing. Cross assignment of peptide ions abundances was performed among the samples and controls using an m/z tolerance of 5 ppm and a retention time tolerance of 42 s. Protein abundances were computed using the median ratio fitting of the unique peptides abundances normalized at the peptide level using the median.
Statistical post-processing
Protein Abundances were loaded into the ProStaR software version 1.16 (http://www.prostar-proteomics.org)32, log2 transformed and associated to their conditions (WT, Lat or Mx2). Proteins with at least 80% of non-missing values in at least one condition of the one vs. one comparison were kept for further statistical analysis. Contaminants and reverse hits were filtered out. Residual missing values were imputed according to their nature33: partially observed values were imputed according to the measured values (Structured Least Squares Algorithm)34, whereas values missing in the entire condition were imputed in a conservative way (approximation of the lower limit of quantification by the 2.5% lower quantile of each replicate intensity distribution).
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository35: the liver dataset is available with the dataset identifier PXD02841636 and the plasma dataset is available with the dataset identifier PXD02855037.
Metabolomics
Three complementary untargeted metabolomics methods based on Ultra-High Performance Liquid Chromatography coupled to High-Resolution Mass Spectrometry (which we will abbreviate as LC-MS in the following for simplicity) were used to ensure a good metabolome coverage. Acquisition of plasma metabolomics data was performed on the three analytical platforms, while metabolomics data from mouse liver was obtained only on two platforms (HILIC and Hypersil C18 as named below), mainly due to the limited sample amounts.
Sample preparation
for further analysis under HILIC and Hypersil C18 conditions: mouse plasma metabolite extraction was performed twice from 50 µl of plasma following methanol-assisted protein precipitation as previously described38. Briefly, a volume of 200 μl of methanol containing internal standards at 3.75 µg/ml (Dimetridazole, 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA), 2-methyl-4-chlorophenoxyacetic acid (MCPA), Dinoseb (Sigma-Aldrich, Saint-Quentin Fallavier, France) was added to the 50 µl of plasma. The resulting samples were then left on ice for 90 min until complete protein precipitation. After a centrifugation step at 20,000 g for 15 min at 4 °C, supernatants were collected and dried under a nitrogen stream using a TurboVap instrument (Thermo Fisher Scientific, Courtaboeuf, France) and stored at −80 °C until analysis. Prior to LC-MS analysis, dried extracts were resuspended in 150 µl of 10 mM ammonium carbonate (pH 10.5) and ACN (40:60, v/v) containing the external standards (mixture of 13C-glucose and 15N-aspartate at 200 µg/ml, ethylmalonic acid at 30 µg/ml, amiloride at 100 µg/ml, prednisone, atropine sulfate and metformin at 10 µg/ml, colchicine and imipramine at 5 µg/ml) for ZIC-pHILIC analysis or water/ACN (95:5, v/v), containing 0.1% FA and the external standards for C18 analysis. A pooled QC sample was obtained by pooling 20 µl of each sample preparation. Aliquots of this QC sample were injected every 5 samples to evaluate potential signal drift of any metabolite, while the external standards added to all samples were used to check for consistency of signal and retention time stability throughout the experiments.
Mouse liver metabolites extraction was performed from ~25 mg of tissue. Samples were resuspended in 170 µl of ultrapure water, and then sonicated 5 times for 10 s using a sonication probe (Vibra Cell, Bioblock Scientific, Illkirch, France). At this step, 20 µl of each sample were withdrawn for further determining the total protein concentration (colorimetric quantification/Pierce BCA Protein Assay Kit, Thermo Fisher Scientific, Courtaboeuf, France). A volume of 350 µl of methanol was added to the remaining 150 µl of tissue lysate. Cell debris were then removed by centrifugation for 15 min at 4 °C and 20,000 g. The resulting samples were then left on ice for 90 min until complete protein precipitation. After a final centrifugation step at 20,000 g for 15 min at 4 °C, supernatants were recovered and split into two equal aliquots for C18 and HILIC analyses. Resulting aliquots were then dried under a stream of nitrogen using a TurboVap instrument (Thermo Fisher Scientific, Courtaboeuf, France) and stored at −80 °C until analysis. Prior to LC-MS analysis, dried extracts were resuspended to reach a fixed protein concentration (equivalent to 20 mg/mL) as described above for plasma-derived metabolites using mobile phase mixtures containing external standards (internal standards were not added for liver metabolomics). After reconstitution, the tubes were vortexed and incubated in an ultrasonic bath for 5 min and then centrifuged at 20,000 g for 15 min at 4 °C. Supernatant was transferred into 0.2 ml vials. QC samples were prepared and analysed as described above for plasma samples.
Sample preparation
for further analysis under HSS T3 C18 conditions: 100 µl of mouse plasma were extracted as follows: samples were slowly thawed on ice at room temperature and protein precipitation was performed by addition of 150 µl of ice-cold methanol. This mixture was vortexed and placed at −20 °C for 30 min. After a 10 min centrifugation (4 °C, 15493 g, Sigma 3-16PK, Fischer Bioblok Scientific), the supernatant was divided into two aliquots, dried completely (EZ2.3 Genevac, Biopharma Technologies France) and stored at −80 °C until further analysis. Just before analysis, 200 µl of injection solvents (water and ACN 50/50 + 0.1% FA) was added to the dried fractions. A pooled QC sample was prepared by mixing 5 µl from each extracted sample and injected every 6 samples.
LC-HRMS Analysis
HILIC and Hypersil C18 analyses: The ultra-high performance liquid chromatographic (UHPLC) separation was performed on a Hypersil GOLD C18 1.9 µm, 2.1 mm × 150 mm column (RP) at 30 °C (Thermo Fisher Scientific, les Ulis, France), and on a Sequant ZICpHILIC 5 µm, 2.1 × 150 mm (HILIC) at 15 °C (Merck, Darmstadt, Germany). All chromatographic systems were equipped with an on-line prefilter (Thermo Fisher Scientific, Courtaboeuf, France). Experimental settings for each LC/MS condition are described below. Mobile phases for the C18 column were 100% water in A and 100% ACN in B, both containing 0.1% FA. Regarding HILIC, phase A consisted of an aqueous buffer of 10 mM ammonium carbonate in water adjusted to pH 10.5 with ammonium hydroxide, whereas pure ACN was used as solvent B. Chromatographic elutions were achieved under gradient conditions as follows: (i) C18-based system: the flow rate was set at 500 µl/min. The elution consisted of an isocratic step of 2 min at 5% phase B, followed by a linear gradient from 5 to 100% of phase B for the next 11 min. These proportions were kept constant for 12.5 min before returning to 5% B for 4.5 min; (ii) HILIC-based system: the flow rate was 200 µl/min. Elution started with an isocratic step of 2 min at 80% B, followed by a linear gradient from 80 to 40% of phase B from 2 to 12 min. The chromatographic system was then rinsed for 5 min at 0% B, and the run ended with an equilibration step of 15 min (80% B).
LC-MS analyses were performed using a U3000 liquid chromatography system coupled to a first generation Exactive mass spectrometer from Thermo Fisher Scientific (Courtaboeuf, France) fitted with an electrospray ionization (ESI) source operated in the positive and negative ion modes. The software interface was Xcalibur (version 2.1; Thermo Fisher Scientific, Courtaboeuf, France). The mass spectrometer was calibrated before each analysis in both ESI polarities using the manufacturer’s predefined methods and recommended calibration mixture provided by the manufacturer (external calibration). The Exactive mass spectrometer was operated with capillary voltage at −3 kV in the negative ionization mode and 5 kV in the positive ionization mode and a capillary temperature set at 280 °C. The sheath gas pressure and the auxiliary gas pressure were set, respectively, at 60 and 10 arbitrary units with nitrogen gas. The mass resolution power of the analyzer was 50,000 (full width at half maximum) at m/z 200, for singly charged ions. The detection was achieved from m/z 85 to 1000 for RP conditions in the positive ionization mode and from m/z 75 to 1000 for HILIC conditions in the negative ionization mode.
LC-HRMS analysis
Acquity HSS T3 C18 analyses: To retain very polar compounds while keeping the elution of less polar metabolites, an HSS T3 column was selected as complementary method39, as this bonding process utilizes a trifunctional C18-alkyl phase bonded at an intermediate 1.6 µmol/m2 ligand density to promote highly polar compounds retention, without extensive retention of less polar components. Metabolic profiles were determined using an U3000 liquid chromatography system (Thermo Fisher Scientific, San Jose, CA, USA) coupled to a high-resolution Bruker Impact ll UHR-QTOF (Bruker Daltonics, Wissembourg, France) equipped with an ESI source. Chromatographic separation was performed on a Waters HSS T3 column (150 × 2.1 mm, 1.8 µm) at 0.4 ml/min, 30 °C and using an injection volume of 5 µl. Mobile phases A and B were water and ACN with 0.1% FA, respectively. The elution gradient was 0% B (2 min), 0–100% B (13 min), 100% B (7 min), 100-0% B (0.1 min) and 0% B (3.9 min for re-equilibration). The mass resolution of the mass spectrometer was 50,000 and mass accuracy ranged from 0.8–2 ppm. Samples were analyzed in the positive and negative ionisation modes (Pos, Neg). Capillary and end plate offset voltages were set at 2,500 V and 500 V for the ESI source. The drying gas temperature was 200 °C and nebulisation gas flow was 10 l/min. Mass spectrum data was acquired in full-scan mode over mass range 50–1000 mass-to-charge ratio (m/z).
The orders of injection of the samples into the LC-HRMS instruments were randomized to ensure that there was no distribution differences between genotypes and between sexes.
Data processing
Raw files were converted to the mzML or mzXML format with the ProteoWizard software40. mzML files were further compressed by using the MS-Numpress tool41. Mass spectra were processed using the xcms R package42 deployed in the Workflow4Metabolomics Galaxy platform (https://workflow4metabolomics.org)43. In particular, the peak detection and quantification was performed with the centWave algorithm44 by using a value of 10 ppm and peak width values of 10,40 (Hypersil C18), 15,90 (HILIC) and 5,20 (Acquity HSS T3 C18). Annotation of the chemical redundancy (adducts, isotopes, fragments) was performed with the CAMERA software45.
Statistical post-processing
Variables whose mean intensity in the biological samples was less than 3 times that of the blanks were discarded. The inclusion of diluted pooled QCs in the Orbitrap datasets enabled further filtering of variables whose linear (Pearson) correlation between the intensity and the inverse of the dilution factor was lower than 0.7. The signal drift observed in the plasma datasets was estimated for each variable by locally weighted (loess) regression and corrected46,47. The reference samples for the regression were the pooled QC samples for the Hypersil C18/HILIC datasets, and the biological samples for the Acquity HSS T3 datasets: for the latter datasets, the normalization based on the pooled QCs was shown to provide only limited correction of the signal drift (as assessed both visually and by the Drift PCA metric described in the Technical Validation section), and the biological samples were therefore used as the reference instead48,49. Variables with a coefficient of variation in the QCs above 30% or greater than the coefficient of variation in the samples were then filtered out. Chemically redundant features (i.e. isotopes, adducts, fragments) were discarded based on the conjunction of three criteria50: Pearson correlation of sample profiles above 0.9, difference between retention times below 6 s, and m/z difference matching a reference list corresponding to isotopes, adducts and fragments at a 0.005 Da tolerance. Finally, intensities were log2 transformed. All steps of this post-processing workflow (Fig. 3) are described in the 2_post_processed vignette from the ProMetIS package27 (Fig. 4), which relies on methods from the phenomis R package (https://github.com/SciDoPhenIA/phenomis).

Post-processing of the ProMetIS datasets. The number of features in each of the 9 datasets is shown. The number of annotated metabolites is indicated in squared brackets. The names of the metabolomics datasets include the reference to the chromatographic column (c18hypersil: Hypersil GOLD C18, hilic: ZIC-pHILIC, and c18acquity: Acquity HSS-T3) and the ionization mode (pos: positive, and neg: negative).

Data and code availability in the ProMetIS package27. The data are available in the extdata folder, which is organized into subfolders corresponding to the successive steps of the analysis (e.g., 1_processed, 2_post_processed, 5_aggregated, etc.). Within each subfolder, the 9 datasets are stored as a triplet of tabular files containing the matrix of intensities (dataMatrix.tsv), the sample metadata (sampleMetadata.tsv) and the variable metadata (variableMetadata.tsv). For each step, a vignette describes the methods used to process the data. As an example, the 2_post_processed vignette details how the datasets were post-processed, between the 1_processed and the 2_post_processed folders (note that only the post-processing of the metabolomics files is described, since the proteomics files were post-processed separately with the ProStaR software). The package thus provides a full access to both data and code, to ensure reproducibility of the results. The tutorial vignette describes how to access the final aggregated data to perform integrative bioinformatics and biostatistics analyses.
Annotation
Metabolite features were annotated according to accurately measured masses and chromatographic retention times by using a local spectral database containing the reference spectra of more than 2,000 authentic standard compounds analyzed in the same analytical conditions, and providing a comprehensive spectral information (i.e. protonated or deprotonated molecules, adducts and in-source fragment ions)38,51. To be identified, ions had to match at least two orthogonal criteria (e.g., accurately measured mass and retention time) to those of an authentic chemical standard analyzed under the same analytical conditions, as proposed by the Metabolomics Standards Initiative52. Additionally, metabolite annotations from the Acquity HSS T3 platform were confirmed by running LC-MS/MS experiments, conducted on the same QC samples, and with the instrument set in the targeted (Multiple Ion Monitoring) acquisition mode. Resulting MS/MS spectra were then manually matched to those included in the in-house spectral database and acquired using different collision energies. For more intelligibility, the annotation confidence was detailed in the datasets regarding the decision criteria (a: accurate mass; b: retention time; c: consistent MS/MS; d: consistent MS/MS with external database).
Data Records
Sample metadata
Throughout the project (including in the data repositories and in the ProMetIS package, the sample names of the 42 mice have been abbreviated to facilitate data manipulation. In particular, a unique project identifier composed of 3 digits is used (ranging from 501 to 862). This identifier is preceded by an uppercase letter referring to the genotype (L = Lat, X = Mx2, W = WT) and followed by a lowercase letter referring to the sex (m = male, f = female): For example, the ‘X501f’ sample comes from the female mouse #501 carrying the Mx2 deletion.
The correspondence between the project identifiers and the reference IDs from the IMPC database is available in the ProMetIS package (in the preclinical subfolders). This table also contains the links to the phenotyping data on the IMPC database. A copy of the table has been included in the Supplementary File 2 for the reader’s convenience (sampleMetadata sheet).
Raw and processed data
Preclinical data are available on the IMPC database at https://www.mousephenotype.org/data/genes/MGI:134229353 and https://www.mousephenotype.org/data/genes/MGI:9724454. MS proteomics data (raw, processed and post-processed files), including peptide data and reference files (readme, search database,.dat files), form a complete submission in the ProteomeXchange repository55. Data were submitted via the PRIDE partner repository under dataset identifiers PXD028416 (liver)36 and PXD028550 (plasma)37. MS metabolomics datasets (raw and processed files) are available for both matrices on the MetaboLights repository56 under the MTBLS1903 identifier57.
Post-processed data
Each post-processed dataset is publicly available in the ProMetIS R package27, as 3 tabular files containing the intensities (dataMatrix), the sample metadata (sampleMetadata) and the variable metadata (variableMetadata) in the extdata folder, either in the 2_post_processed subfolder or in the 5a/b_aggregated_LAT/MX2 subfolders (the latter further include results from statistical analyzes as additional sample and variable metadata). These files may be conveniently opened with any spreadsheet editor. They may be imported as an ExpressionSet58 or MultipleDataSet59 for further single- or multi-omics analysis in R (see for instance the tutorial vignette from the ProMetIS package). Alternatively, they may be analyzed by using the Galaxy tools from the Workflow4Metabolomics platform43,60. Importantly, the post-processing steps used to generate the metabolomics files are described in the 2_post_processed vignette from the ProMetIS package to meet the Findable, Accessible, Interoperable and Reusable FAIR guidelines28. Those post-processed datasets (annotated variables only) are also provided in the Supplementary File 2 for the reader’s convenience (with one sheet per dataset).
Technical Validation
The ProMetIS dataset was generated by the four French Infrastructures for Mouse Phenogenomics (Phenomin-ICS; http://www.phenomin.fr/en-us), Proteomics (ProFI; http://www.profiproteomics.fr), Metabolomics and Fluxomics (MetaboHUB; https://www.metabohub.fr/home.html) and Bioinformatics (IFB; https://www.france-bioinformatique.fr) which meet the international standards for optimal quality, reproducibility and accessibility of the data.
Preclinical data
Routine procedures to optimize the quality of phenotyping data at the Phenomin-ICS infrastructure include i) standardized pipelines and experiments, ii) automated flagging of outlier variables by comparing the WT mice from the project to all other WT mice from the same genetic background previously generated by the platform (as described below), and iii) manager data review and validation.
For each quantitative variable, a reference range encompassing 95% of the values from all WT male and female mice with the same genetic background in the Phenomin-ICS database is computed (rr95). For the variables which follow a normal distribution (either directly or after an inverse, logarithm or square root transformation), as assessed with the Shapiro-Wilk test, the rr95 is set to the average ± 2 × standard deviation (this is the case for about two thirds of the variables). For the other variables, the rr95 bounds are defined as the 2.5th and 97.5th percentiles of the values. In all cases, a minimal number of 120 animals is recommended by the EP28 standard from the Clinical and Laboratory Standards Institute (CLSI; https://clsi.org) to ensure stable lower and upper reference limits. Variables with an average value in the WT mice of the project falling out of the rr95 for males or females are discarded.
Here, the rr95 was computed from 1,112 WT mice (573 males and 539 females). The transformation applied to compute the rr95 has been included in the metadata. For all 200 preclinical variables, the 15 WT mice from the ProMetIS project had average values within the rr95 in males and females, thus confirming the good stability and reproducibility of the measures performed in this study (Fig. 5a). The quality of the preclinical phenotyping was also assessed at the multivariate level by Principal Component Analysis: as expected, ProMetIS mice were located within the cloud of all Phenomin-ICS mice on the score plot (Fig. 5b).

Quality control of the preclinical dataset assessed on the WT mice. (a) Example of the values of the parameter “body weight at week 7” in the WT mice from the ProMetIS dataset compared to the other Phenomin-ICS WT mice. Blue (respectively, orange) rectangles correspond to the rr95 reference range for males (respectively, females). The batch from the 15 WT mice from the ProMetIS study is indicated by an arrow. (b) Principal component analysis of all WT mice from the Phenomin-ICS database. The mice from the ProMetIS dataset are colored in red.
Proteomics data
The proteomics study of mice liver (respectively, plasma) samples based on tryptic peptides analyzed by nanoLC-MS resulted in the raw quantification of 21,809 (respectively, 6,501) peptides and 2,468 (respectively, 621) proteins at the end of the data processing by the Proline software. Three types of quality control strategies were used throughout the analytical sequence. Analytical conditions were optimised for each of the two tissues since the study was designed for the comparison of mutants vs wild-type samples within each tissue, taken individually. The present experimental design does not therefore allow the direct comparison of protein absolute quantification between the plasma and liver contents. First, an internal standard was spiked in equal amounts in each biological sample and to check the stability of the LC and MS systems. This verification was done post-acquisition of the sequence, through extraction of the retention times (RT) and the raw intensity data associated with the spiked peptides from the quantitative output of the Proline software (Figure S1 of the Supplementary File 1). This spiked-in standard is composed of eleven pure synthetic peptides (iRT kit, Biognosys, # Ki-3002-1). Figure 6 (left) and Figure S1 of the Supplementary File 1 illustrate the reproducibility of the features (retention time and raw intensities, respectively) associated with these iRT peptides across the LC-MS runs of the dataset. In particular, the median CV of intensities across all liver (respectively, plasma) samples injections was 40% (respectively, 31%; Figure S1 of the Supplementary File 1). Second, an external standard of close composition to that of the biological samples themselves was regularly injected during the acquisition sequence. It was prepared by pooling a small aliquot of all initial samples. An LC-MS run of this pool standard was acquired every 4 or 5 analytical runs during the sequence (n = 10 pools for liver samples, n = 8 pools for plasma samples). These runs were included, together with those of the samples, in the data processing performed with the Proline software. The identification metrics extracted from the data of this pooled external standard (i.e. number of proteins, peptides, MS/MS scans; Figure S2, left, and Figure S3, left, of the Supplementary File 1, as well as the CVs of the raw protein intensities, Fig. 6, right, and the correlation between the samples, Figure S4 of the Supplementary File 1) show high repeatability of the results. Third, quality control of the biological datasets was assessed by plotting the same identification and quantitative metrics across all biological samples (Figure S2, right, Figure S3, right, of the Supplementary File 1, and Fig. 6, right). No outlier was identified in the cohorts. The variability of these metrics was close to the one of the pools and was used to assess the global quality of the datasets, excluding any variability related to the composition of the biological samples.

Quality control of the LC-MS proteomics analytical sequences in the liver (top) and plasma (bottom). Left: An internal standard consisting of 11 iRT peptides (iRT kit; Biognosys) was spiked in all samples. Retention times of the 11 spiked in peptides from the iRT kit (Biognosys) are plotted in the chronological order of data acquisition. Mean standard deviation across all liver (respectively, plasma) samples injections are 27 s (respectively, 13.5 s). The sequences of the peptides are: P1: LGGNEQVTR, P2: YILAGVENSK, P3: GTFIIDPGGVIR, P4: GTFIIDPAAVIR, P5: GAGSSEPVTGLDAK, P6: TPVISGGPYEYR, P7: VEATFGVDESNAK, P8: TPVITGAPYEYR, P9: DGLDAASYYAPVR, P10: ADVTPADFSEWSK, P11: LFLQFGAQGSPFLK. Right: CVs of the raw protein intensities per sample type.
Metabolomics data
The quality of the metabolomics datasets was investigated after the post-processing step (including the signal drift correction but not the final feature filtering on the pooled QC CVs), as a validation of the data before future statistical studies.
The stability of the signal detection and quantification along the sequence of injection was first assessed on the metabolite internal and external standards which have been spiked in all crude plasma samples before metabolite extraction (internal standards), or in the final extracts just before injection into the LC-HRMS (external standards)38,61,62. Of note, the mixture of 13 external standards is used as an “universal” standard solution in the laboratory, i.e. whatever the sample to be analyzed (e.g., biofluid, tissue, bacteria, or fecal samples) and LC-MS conditions, which implies that not all the chemicals are efficiently detected and thus quantified in every type of metabolite extract or analytical conditions (e.g., positive vs negative ionisation). For instance, 9 and 4 external standards were detected in plasma extracts analyzed under c18hypersil_pos and hilic_neg conditions, respectively. Although all those standard compounds proved efficiently detected in liver extracts when analyzed under hilic_neg conditions, c18hypersil_pos analysis detected 7 among the 9 external standards potentially due to stronger matrix effects occurring with liver extracts. For all detected standards, the average coefficient of variation was 2% (standard deviation: 1%; Figure S5 of the Supplementary File 1). The few samples with a z-score absolute value above 3 for one of the metabolite standards were manually validated: no consistent outlier pattern across all spiked standards nor any outlier location on the PCA score plot were observed for any of those samples.
We further used a set of five metrics to assess the global quality of all six metabolomics datasets: the first two metrics assess the absence of any analytical drift between the samples by using univariate and multivariate approaches. The remaining three scores focus on the quality of the pooled QC samples: we first define a scoring of the QC spread on the PCA score plot, and then include two metrics corresponding to the coefficient of variation (CV)47,48 and the intraclass correlation coefficient (ICC)63. Each metric, ranging from 0 to 100 is computed as follows (a score of 100 corresponds to the highest quality):
-
1.
Drift Spearman (proportion of features which are not significantly correlated with the injection order): \(\left(1-\frac{{p}_{correlated}}{{p}_{total}}\right)\times 100\), where pcorrelated is the number of features which are correlated to the injection order (False Discovery Rate: 5%) and ptotal is the total number of features
-
2.
Drift PCA (proportion of variance of the injection order which is not explained by the 3 first PCA components): (1−cos2α) × 100, where a is the angle between the direction of the injection order and its projection on the PCA hyperspace of the 3 first components; a value of 100 (i.e. cosα = 0) corresponds to the absence of correlation with the injection order (the direction of the injection order gradient is orthogonal to the PCA 3D hyperspace)
-
3.
QC spread (spread of the pooled QC samples in the PCA 3D hyperspace): \(\left(1-\frac{max{\rm{(}}{d}_{QC}{\rm{)}}}{max{\rm{(}}{d}_{sample}{\rm{)}}}\right)\times 100\), where max(dQC) is the Mahalanobis distance to the center of the farthest pooled QC sample in the PCA 3D hyperspace and max(dsample) is the distance of the farthest biological sample
-
4.
QC CV (coefficient of variation of the pooled QC intensities): percentage of features with a coefficient of variation of the intensities in the QCs inferior to 30%47,48: \(\frac{{p}_{CV\le 0.3}}{{p}_{total}}\times 100\), where pCV ≤ 0.3 is the number of features with an intensity CV of less than 30% in the pooled QC; recently, Zhang et al. (2020) proposed to further display the cumulative percentage of compounds as a function of CV for a more detailed overview of the dataset quality63. The corresponding graphic is therefore provided as the Figure S7 of the Supplementary File 1
-
5.
QC ICC (intraclass correlation coefficient, ICC, between the pooled QC intensities at the most probable abundance63): ICCmost probable abundance × 100, the use of the ICC between the pooled QC has been recently proposed as a metric to assess the reliability of the measurements63. The cumulative ICCs as a function of the intensities are shown in the Figure S8 of the Supplementary File 1, and the ICC value at the intensity bin containing most compounds (most probable abundance) is used as the quality metric63. The icc function from the irr package64 was used with the “twoway” random effect model, “agreement”, and “single” parameters.
High scores were obtained for all datasets, with an average value of the metrics ranging between 84 and 94% (for the plasma_c18acquity_pos and plasma_hilic_neg datasets, respectively; Table 1 and Fig. 7). The normalization based on pooled QCs (respectively, on samples) was necessary to achieve a correction of the signal drift for the plasma_c18hypersil/hilic datasets (respectively, the plasma_c18acquity datasets, Figure S6 of the Supplementary File 1), as assessed by the Drift Spearman and Drift PCA metrics scores above 90%. Furthermore, the three QC metrics of the datasets were high (QC spread, QC CV, QC ICC), even in the absence of signal drift correction or after a normalization based on the samples, with an average value of 83% (min-max: 58–99%). The curves showing the cumulative percentage of compounds as a function of the CVs in the pooled QC samples were similar to those obtained by Zhang et al.63 with a repeated injection of a single serum sample (Figure S7 of the Supplementary File 1): in all metabolomics datasets, a minimum of 79% of features had a CV below 30%. The cumulative ICC as a function of the intensity in the pooled QCs was similar to those from Zhang et al.63, with an ICC value at the most probable intensity between 71% and 89%, except for the plasma_c18acquity datasets which had slightly lower values (58% and 65%), confirming that although the pooled QCs were less reliable in these latter datasets, the overall quality of all metabolomics datasets was high (Figure S8 of the Supplementary File 1). Finally, the distribution of CVs in the biological samples were similar for all genotypes (Figure S9 of the Supplementary File 1), as observed in proteomics (Fig. 6 right).

Quality control of the metabolomics datasets. (a) Scores of the datasets according to five metrics visualized as a radar plot. The two “Drift” metrics assess the absence of unwanted variation due to a signal drift by univariate and multivariate approaches, whereas the three “QC” metrics evaluate the quality of the pooled QC samples. CV: coefficient of variation; ICC: intraclass correlation coefficient. (b) PCA score plots displaying the samples and pooled QCs with a color gradient according to the injection order. QC: pooled quality control sample; s: biological sample.
Sex-specific validation
Individual features with significant difference in means between males and females were detected in all ProMetIS datasets, as it is often observed in phenotyping studies (from 14% to 40% of the total number of features, depending on the dataset; limma test, 5% threshold, Benjamini and Hochberg correction of the False Discovery Rate). We therefore checked the absence of any systematic bias related to sex on feature intensities, CVs and imputation (Figures S10, S11 and S12, respectively, of the Supplementary File 1).
Usage Notes
The tutorial vignette from the ProMetIS package27 describes in detail how to load the data and provides examples of single omics and integrative data analysis of the datasets with R packages, such as univariate hypothesis testing (phenomis; https://github.com/SciDoPhenIA/phenomis), orthogonal partial least-square – discriminant analysis (ropls)48, feature selection (biosigner)65, sparse multi-block PLS-DA modeling (mixOmics)66.
Alternatively, the proteomics and metabolomics data may also be readily loaded and analyzed online by using the Workflow4Metabolomics43 and ProteoRE67 platforms. These resources are based on the Galaxy environment to facilitate workflow management68, and include many modules for statistical analysis and annotation60,69.
Code availability
The dataset and the source code for the data analysis are publicly available as the ProMetIS R package27 on GitHub (https://github.com/IFB-ElixirFr/ProMetIS). The package includes several vignettes describing the pre-processing of the data as well as the tables and figures from this article (6_article_data).
References
EUMODIC Consortium. et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat. Genet. 47, 969–978 (2015).
The International Mouse Phenotyping Consortium. et al. High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514 (2016).
Meehan, T. F. et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 49, 1231 (2017).
Aebersold, R. & Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 (2016).
Patti, G. J., Yanes, O. & Siuzdak, G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 13, 263–269 (2012).
Rolin, D. Metabolomics coming of age with its technological diversity. Adv. Bot. Res. 67, 2–693 (2013).
Piazza, I. et al. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 172, 358–372.e23 (2018).
Parker, B. L. et al. An integrative systems genetic analysis of mammalian lipid metabolism. Nature 567, 187–193 (2019).
Shen, B. et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 182, 59–72.e15 (2020).
Ghaemi, M. S. et al. Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy. Bioinformatics 35, 95–103 (2018).
Webb-Robertson, B. J. M. et al. A bayesian integration model of high-throughput proteomics and metabolomics data for improved early detection of microbial infections. Pac. Symp. Biocomput. 19, 451–463 (2009).
Fischer, R., Bowness, P. & Kessler, B. M. Two birds with one stone: Doing metabolomics with your proteomics kit. Proteomics 13, 3371–3386 (2013).
Martens, L. et al. mzML - a Community Standard for Mass Spectrometry Data. Mol. Cell. Proteomics 10 (2011).
Griss, J. et al. The mzTab Data Exchange Format: Communicating Mass-spectrometry-based Proteomics and Metabolomics Experimental Results to a Wider Audience. Mol. Cell. Proteomics 13, 2765–2775 (2014).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Kenar, E. et al. Automated Label-free Quantification of Metabolites from Liquid Chromatography–Mass Spectrometry Data. Mol. Cell. Proteomics 13, 348–359 (2014).
Gatto, L., Gibb, S. & Rainer, J. MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J. Proteome Res. 20, 1063–1069 (2021).
Perez‐Riverol, Y. & Moreno, P. Scalable Data Analysis in Proteomics and Metabolomics Using BioContainers and Workflows Engines. PROTEOMICS 20, 1900147 (2020).
Coman, C. et al. Simultaneous Metabolite, Protein, Lipid Extraction (SIMPLEX): A Combinatorial Multimolecular Omics Approach for Systems Biology. Mol. Cell. Proteomics 15, 1435–1466 (2016).
Blum, B. C., Mousavi, F. & Emili, A. Single-platform ‘multi-omic’ profiling: unified mass spectrometry and computational workflows for integrative proteomics–metabolomics analysis. Mol. Omics 14, 307–319 (2018).
Zougman, A., Wilson, J. P., Roberts, L. D. & Banks, R. E. Detergent-Free Simultaneous Sample Preparation Method for Proteomics and Metabolomics. J. Proteome Res. 19, 2838–2844 (2020).
Brown, S. D. M. et al. High-throughput mouse phenomics for characterizing mammalian gene function. Nat. Rev. Genet. 19, 357–370 (2018).
Roncagalli, R., Mingueneau, M., Grégoire, C., Malissen, M. & Malissen, B. LAT signaling pathology: an “autoimmune” condition without T cell self-reactivity. Trends Immunol. 31, 253–259 (2010).
Loviglio, M. N. et al. The immune signaling adaptor LAT contributes to the neuroanatomical phenotype of 16p11.2 BP2-BP3 CNVs. Am. J. Hum. Genet. 101, 564–577 (2017).
Muñiz Moreno, M. et al. Modeling Down syndrome in animals from the early stage to the 4.0 models and next. in Progress in Brain Research vol. 251, 91–143 (Elsevier, 2020).
Barupal, D. K. et al. A Comprehensive Plasma Metabolomics Dataset for a Cohort of Mouse Knockouts within the International Mouse Phenotyping Consortium. Metabolites 9, 101 (2019).
Imbert, A. & Thévenot, E. The ProMetIS R package: Multi-omics phenotyping of the LAT and MX2 knockout mice. Zenodo https://doi.org/10.5281/zenodo.5615546 (2021).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Brown, S. D. M. & Moore, M. W. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mamm. Genome 23, 632–640 (2012).
Simon, M. M. et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 14, R82 (2013).
Bouyssié, D. et al. Proline: an efficient and user-friendly software suite for large-scale proteomics. Bioinformatics 36, 3148–3155 (2020).
Wieczorek, S. et al. DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 33, 135–136 (2017).
Lazar, C., Gatto, L., Ferro, M., Bruley, C. & Burger, T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 15, 1116–1125 (2016).
Gianetto, Q. G., Wieczorek, S., Couté, Y. & Burger, T. A peptide-level multiple imputation strategy accounting for the different natures of missing values in proteomics data. Preprint at https://doi.org/10.1101/2020.05.29.122770 (2020).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Rompais, M., Cianferani, S. & Carapito, C. ProMetIS: deep phenotyping of mouse models by proteomics and metabolomics. PRIDE https://identifiers.org/pride.project:PXD028416 (2020).
Mouton-Barbosa, E., Schiltz, O. & Gonzalez de Peredo, A. ProMetIS: deep phenotyping of mouse models by proteomics and metabolomics. PRIDE https://identifiers.org/pride.project:PXD028550 (2020).
Boudah, S. et al. Annotation of the human serum metabolome by coupling three liquid chromatography methods to high-resolution mass spectrometry. J. Chromatogr. B 966, 34–47 (2014).
Pereira, H., Martin, J.-F., Joly, C., Sébédio, J.-L. & Pujos-Guillot, E. Development and validation of a UPLC/MS method for a nutritional metabolomic study of human plasma. Metabolomics 6, 207–218 (2010).
Kessner, D., Chambers, M., Burke, R., Agus, D. & Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008).
Teleman, J. et al. Numerical Compression Schemes for Proteomics Mass Spectrometry Data. Mol. Cell. Proteomics 13, 1537–1542 (2014).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Giacomoni, F. et al. Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics 31, 1493–1495 (2015).
Tautenhahn, R., Bottcher, C. & Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9, 504 (2008).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 84, 283–289 (2012).
Cleveland, W. S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 74, 829–836 (1979).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 6, 1060–1083 (2011).
Thévenot, E. A., Roux, A., Xu, Y., Ezan, E. & Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 14, 3322–3335 (2015).
Wehrens, R. et al. Improved batch correction in untargeted MS-based metabolomics. Metabolomics 12, 1–12 (2016).
Monnerie et al. Analytic Correlation Filtration: a new tool to reduce analytical complexity of metabolomic datasets. Metabolites 9, 250 (2019).
Roux, A. et al. Annotation of the Human Adult Urinary Metabolome and Metabolite Identification Using Ultra High Performance Liquid Chromatography Coupled to a Linear Quadrupole Ion Trap-Orbitrap Mass Spectrometer. Anal. Chem. 84, 6429–6437 (2012).
Sumner, L. et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221 (2007).
Lat: linker for activation of T cells. Mouse Genome Informatics Database https://identifiers.org/MGI:1342293 (2018).
Mx2: MX dynamin-like GTPase 2. Mouse Genome Informatics Database https://identifiers.org/MGI:97244 (2018).
Vizcaíno, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014).
Haug, K. et al. MetaboLights: an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 41, D781–D786 (2013).
Thévenot, E. A., Castelli, F., Pujos-Guillot, E. & Fenaille, F. MTBLS1903: ProMetIS: deep phenotyping of knock-out mice by proteomics and metabolomics. MetaboLights https://identifiers.org/metabolights:MTBLS1903 (2021).
Huber, W. et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121 (2015).
Hernandez-Ferrer, C., Ruiz-Arenas, C., Beltran-Gomila, A. & Gonzalez, J. R. MultiDataSet: an R package for encapsulating multiple data sets with application to omic data integration. BMC Bioinformatics 18, 36 (2016).
Guitton, Y. et al. Create, run, share, publish, and reference your LC-MS, FIA-MS, GC-MS, and NMR data analysis workflows with the Workflow4Metabolomics 3.0 Galaxy online infrastructure for metabolomics. Int. J. Biochem. Cell Biol. 93, 89–101 (2017).
Broadhurst, D. et al. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 14, 72 (2018).
Dudzik, D., Barbas-Bernardos, C., Garcia, A. & Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. Rev. Issue 2017 147, 149–173 (2018).
Zhang, X., Dong, J. & Raftery, D. Five Easy Metrics of Data Quality for LC–MS-Based Global Metabolomics. Anal. Chem. 92, 12925–12933 (2020).
Gamer, M., Lemon, J., Fellows, I. & Singh, P. irr: Various Coefficients of Interrater Reliability and Agreement. https://CRAN.R-project.org/package=irr (2019).
Rinaudo, P., Boudah, S., Junot, C. & Thévenot, E. A. biosigner: a new method for the discovery of significant molecular signatures from omics data. Front. Mol. Biosci. 3 (2016).
Rohart, F., Gautier, B., Singh, A. & Le Cao, K.-A. mixOmics: An R package for omics feature selection and multiple data integration. PLOS Comput. Biol. 13, e1005752 (2017).
Vandenbrouck, Y., Christiany, D., Combes, F., Loux, V. & Brun, V. Bioinformatics Tools and Workflow to Select Blood Biomarkers for Early Cancer Diagnosis: An Application to Pancreatic Cancer. PROTEOMICS 19, 1800489 (2019).
Goecks, J., Nekrutenko, A. & Taylor, J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86 (2010).
Combes, F., Loux, V. & Vandenbrouck, Y. GO Enrichment Analysis for Differential Proteomics Using ProteoRE. in Proteomics Data Analysis (ed. Cecconi, D.) vol. 2361, 179–196 (Springer US, 2021).
Acknowledgements
We thank David Vallenet, Olivier Sand and Jacques van Helden for the scientific support of the project. We thank the PRIDE and MetaboLights teams for their help during the submission and curation of the datasets. We thank both anonymous reviewers for their constructive comments that helped to further improve the quality of the presented data. This work was funded by the four French Infrastructures for Mouse PhenoGenomics (Phenomin-ICS, ANR-10-INBS-0007), Proteomics (ProFI, ANR-10-INBS-0008), Metabolomics (MetaboHUB, ANR-11-INBS-0010), and Bioinformatics (IFB, ANR-11-INBS-0013).
Author information
Authors and Affiliations
Contributions
M.S., T.S. and Y.H. generated the preclinical data; S.L., T.S., Y.H., A.I. and E.T. analyzed the preclinical data; M.R., A.H., E.M.B., C.C. and A.G. generated the proteomics data; M.R., E.M.B., C.C., A.G., Y.V., T.B., A.I. and E.T. analyzed the proteomics data; E.C.V. and C.J. generated the metabolomics data; F.C., M.B.B., P.R., S.O., F.F., E.P.G., C.J., A.I. and E.T. analyzed the metabolomics data; A.I. and E.T. developed the software package; A.I., E.T., Y.H., F.F., E.P.G., M.R., E.M.B., C.C., A.G., M.F., Y.V., C.M., M.S. and S.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Imbert, A., Rompais, M., Selloum, M. et al. ProMetIS, deep phenotyping of mouse models by combined proteomics and metabolomics analysis. Sci Data 8, 311 (2021). https://doi.org/10.1038/s41597-021-01095-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-01095-3
