
Abstract
Prokaryoplankton genomes from the deep marine sediments are less explored compared to shallow shore sediments. The Gulfs of Kathiawar peninsula experience varied currents and inputs from different on-shore activities. Any perturbations would directly influence the microbiome and their normal homeostasis. Advancements in reconstructing genomes from metagenomes allows us to understand the role of individual unculturable microbes in ecological niches like the Gulf sediments. Here, we report 309 bacterial and archaeal genomes assembled from metagenomics data of deep sediments from sites in the Gulf of Khambhat and Gulf of Kutch as well as a sample from the Arabian Sea. Phylogenomics classified them into 5 archaeal and 18 bacterial phyla. The genomes will facilitate understanding of the physiology, adaptation and impact of on-shore anthropogenic activities on the deep sediment microbes.
Measurement(s) | marine metagenome • sequence_assembly |
Technology Type(s) | DNA sequencing • sequence assembly process • Binning clustering method |
Factor Type(s) | gulf |
Sample Characteristic - Environment | marine biome • deep marine sediment |
Sample Characteristic - Location | Gulf of Kutch • Gulf of Khambhat • Arabian Sea |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.14445387
Similar content being viewed by others

Background & Summary
Marine microbiome is considered as the largest environment on earth which has many secrets concealed into it1,2. Many marine microbes play a key role in biogeochemical cycles. However, high proportions of microbes remain uncultured in vitro3 and so instead of analysing the microbes individually, cultivation-independent genome-level characterization methods notably single-cell genomics and metagenomics are frequently being applied for microbiome analysis4. Amplicon sequencing based cultivation-independent studies are enriching the microbial diversity knowledge of various hitherto less studied environmental niche, specifically within the marine resources. However, amplicon analysis is just a preliminary step in metagenomics as it focuses only on one gene for the community diversity assessment.
With the view of studying the marine microbial community for determination of its composition in terms of diversity as well as function, whole metagenomics has become the preferred approach. Recently, it has been realized that the actual understanding of metagenomics data can be obtained by individual genome binning, which eventually also enhances the microbial genome database5. This requires use of various complex computational algorithms including those relying on previous data findings viz., the supervised classifiers and the unsupervised classifiers that rely on sequence specific features like the GC content, k-mer frequency and coverage estimation for binning the genomes. Most of the recently developed tools for binning include a combined approach of both the algorithms6. Binning aids in revealing the link between the potential functional genes in a given microbiome to its taxonomy.
The unique properties of the Gulfs of Kathiawar Peninsula like extreme tidal variations, different sediment texture and physicochemical variations make them an ideal place for studying the microbial diversity. Varied onshore anthropogenic activities may have imparted unique features to the microflora of the Gulfs. Study of microbial diversity and functions in the mentioned Gulfs have largely been focused on cultivation based approaches and very few molecular studies have been conducted on the shore sediments. Additionally, the presence of several on-shore industries like fertilizer, chemicals, oil refineries, power plants and ASSBRY (Alang Ship Breaking Yard) may have also influenced the deeper sediment microbiome leading to their variable gene profile7. Our previous insights into the pelagic sediment resistome profile by metagenomics approach have shown that the deeper sediments, earlier thought to be primeval are actually hosting microbes with a concerning number of resistance genes7,8. This acted as a propeller to the present study wherein we tried to look deeper into the metagenomics data of the samples collected from the Gulfs of Kathiawar Peninsula and a sample from the Arabian Sea by sorting individual prokaryoplankton genomes from the data using the binning approach.
We successfully reconstructed 309 Metagenome Assembled Genomes (MAGs) from the nine sediment metagenomics sequences (Table 1) from Gulf of Khambhat (GOC), Gulf of Kutch (GOK) and Arabian Sea (A) by differential coverage approach and considering the GC percent and tetranucleotide frequencies. Out of the 309 MAGs, 39 were archaeal genomes (Online-only Table 1) and 270 were bacterial genomes (Online-only Table 2). Seventy-one were high quality drafts with a completeness of ≥90% and contamination <10%, 120 were medium quality (completeness: 70–90%, contamination: <10%) and the remaining 118 were draft genomes with a final completeness of >50%. The distribution of the bins as per the MIMAG quality standards9 is described in Table 2. To the best of our knowledge, this is the first report of multiple MAGs from the studied sites.
Single nucleotide polymorphisms were correlated to quality of bins to understand the influence of strain heterogeneity on the fragmentation of the MAGs (Fig. 1). Phylogenomic analysis revealed that the archaeal populations were quite different in two Gulfs, with GOC bins (n = 15) encompassing 3 major phyla: Thaumarchaeota and Aenigmarchaeota from the DPANN superphylum andBathyarchaeota. The GOK genomes (n = 24) were falling under the Bathyarchaeota, Thaumarchaeota, Euryarchaeota and the Korarchaeota phyla (Figs. 2 and 3). Based on the community profile assessment of the samples by considering all the reads, the above mentioned archaeal phyla represented <3% of the total microbial population at each sample site. Majority of the phyla were those reported earlier in the marine and estuarine environments, with most having few or no cultured representatives10,11. The observed genomes like Thaumarchaeota have been reported to be nitrifiers in the sediment niche, thus, the insights into their gene content will provide details on the functional significance of the archaea in the respective sample site. Genomes from Thaumarchaeota were recovered from both the sites (Fig. 2). Nevertheless, the difference in the populations observed in two Gulfs can also be studied based on the predicted roles of the genomes and correlation with the niche properties.

Single nucleotide polymorphisms (SNPs) were called for the MAGs reported here and compared with (a) quality, (b) sample site and (c) N50. The values were plotted as box plot with Min/Max whiskers and line in the middle corresponding to the mean value.

The distribution of MAGs across archaeal and bacterial phyla in the studied sites. Four MAGs that were classified only up to the domain level have not been depicted here.

Phylogenetic tree of the archaeal MAGs. Validity of the tree is indicated by filled black circles, size indicates bootstrap between 80 to 100%. The tree was rooted with the Aenigmarchaeota of the DPANN superphylum4.
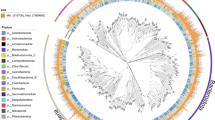
Among the bacterial members, five phyla were commonly observed between the Gulfs viz., Proteobacteria, Zixibacteria, Gemmatimonadetes, Dadabacteria and Planctomycetes (Figs. 2 and 4). Among the common bacterial phyla, Proteobacteria majorly comprised of Gammaproteobacteria members which are the most abundant reported bacteria in the marine sediments and have been reported to perform versatile roles including metabolite production, hydrocarbon degradation, acetate assimilation and many more12,13. Zixibacteria and Dadabacteria MAGs have been reported from marine environments as an evolutionary phyla and these have been observed to play role in the nutrient cycling of the niche14,15. Apart from these, few genomes in GOC encompassed Bacteroidetes, FCB superphylum, Armatimonadates, Acidobacteria, Chloroflexi and Aminicenantes phyla; while those in GOK were falling under Actinobacteria, KSB1, Saccharibacteria (TM7), Nitrospinae, Caldithrix, Verrucomicrobia and Balneolaeota. Species belonging to Nitrospinae are reported to be exclusively abundant in marine niche, where they play a role in nitrite oxidization, as well as these are ubiquitously observed in sites demanding thermoprotection16,17. Community profiling of the samples by considering all the reads revealed that the MAGs identified within Proteobacteria (>40%) and Chloroflexi (~15%) phyla represented a substantial population, while rest of the MAGs corresponded to 0.01% to 5% of the total microbial community at each sample site (details in Supplementary Table 1a and b).

Bacterial clades having ≥10 MAGs classified as the same level are collapsed and represented by triangles, size of which is proportional to the number of genomes collapsed in the taxa level which is also mentioned in the parentheses. Validity of the tree is indicated by filled black circles, size indicates bootstrap between 80 to 100%. The Akkermansiaceae (phylum: Verrucomicrobia) was arbitrarily taken as the root, the tree may be considered as unrooted34.
The genomic bins described here would prominently enhance the repertoire of microbial genomic information from the Gulfs of Kathiawar Peninsula. It will also provide the insights for better understanding the effects of on-shore activities on the microbiome of deeper sediment in the Gulfs. In the long term, the data will fortify further applications of the genomic information for 1) understanding the microbes involved in the marine nutrient cycling, 2) open gates for bioprospection of novel thermophilic and halophilic enzymes and 3) allow understanding of microbial-host and microbial-niche interactions as the phylum distribution reflects the variability across the 2 Gulfs under study.
Methods
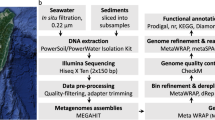
Sample collection and whole metagenome sequencing
The sediment samples were collected and sequenced for whole metagenomics using Illumina HiSeq platform as described earlier7,8. In brief, one-meter-long sediment cores were collected from 9 locations across the 2 Gulfs, Gujarat state and open Arabian Sea by sailing through boats. The cores were maintained in cold storage and processed by cutting into halves without disturbing the sediments. 10 cm of sediment from top, middle and bottom of the core each was distributed into 3 sterile 50 ml collection containers. They were further used for assessment of physicochemical properties, metagenomic DNA isolation and culturing purpose. DNA was isolated in multiplicates to reach desired quantity using the MoBio Power Soil DNA isolation Kit (Qiagen, Germany). The DNA from each core section was pooled in equimolar concentrations for whole metagenomics sequencing using HiSeq 4000 (Macrogen Inc., Korea). No internal reference or control was used during the sequencing. The sequences were quality filtered for adaptor removal and a minimum quality score of 20.
Metagenomics assembly
The quality filtered reads of four GOK samples were used for pooled assembly using CLC Genomics Workbench v11.0 with default parameters except a k-mer size of 31 and a minimum contig length of 1 Kb, which resulted into 478 Mb of assembled data. Similarly, the four samples of GOC along with the A sample were included in another set of pooled assembly of 779 Mb. The raw reads from each of the individual nine samples were mapped against the two assemblies for coverage estimation using CLC Genomics Workbench. The coverage and the BAM files were obtained for further binning process.
Genome binning
Metagenome assembled genomes (MAGs) were binned from both the assemblies using Maxbin v2.06 using the full reference marker set of genes for bacteria and archaea. More than 900 bins were initially obtained from the pooled datasets. The quality of bins was checked by CheckM v1.1.0 using the lineage-specific workflow and the bins were assessed based on its completeness and contamination values18. Contigs with outlier values for the GC percentage and tetranucleotide frequency were removed from the bins for lowering the contamination levels. The assessed bins were further refined using RefineM v0.0.22 by individual genomic properties, taxonomy and SSU based approaches. Further, the individual output of the genomic properties was used as input for the other methods, viz., output bins from the genomic property refined program was further filtered by taxonomic method and so on. The refined bins were re-assessed using CheckM v1.1.018. It was observed that the refinement using the genomic properties which screens for any outlier contigs/scaffoled in a MAG in terms of GC percent, tetranuceotide frequency and coverage did improve the bins in terms of their completeness. While, the taxa based refinement gave an overall improvement of ~2% for few bins and a reduction in the contamination by removal of duplicate or miss-assigned single copy gene encoding contigs. SSU based refinement had no major impact on the MAGs in the study. The bins were then sorted into high quality, medium quality, draft and/or low quality genomes. Out of >900 bins, 309 bins that were falling up to the draft genome category were checked for taxonomic classification using the GTDB-tk v0.3.3 classifier19. However, for final submission as suggested by NCBI team, few of the NCBI taxonomic synonyms from GTDB-tk classification were considered. The strain names in the nomenclature were assigned as “sample number of the mapped reads – pooled assembly against which the reads were mapped followed by the number of bin from the mapped sample”, as an example for the strain CS3-K071, CS3 indicates Gulf of Khambhat/Cambay Sample 3 which was mapped against the pooled assembly of Kutch samples and 071 is the bin number from the total bins generated from this mapping. The bins were also submitted to RAST v2.0 for annotation and the number of Protein Encoding Genes (PEGs) for each MAG were inferred from the same for preliminary functional assessment prior to NCBI submission20.
SNP estimation of the MAGs
SNPs were called for each MAG (n = 275, bins generated from all nine samples as a pool were omitted from SNP analysis) (Supplementary Table 1a and b) to assess their genetic diversity as described earlier21. For the same, a database of MAGs from each of the nine samples was computed using Bowtie2 v.2.3.4.122 for aligning the respective metagenomic reads. The SNP/Kbps were compared with the quality, sample site and N50 and of the MAGs. All the plots were computed using GraphPad Prism v8.4.1 for Windows23.
Phylogenomic tree construction
The archaeal and bacterial trees were inferred using the insert genome set into species tree app in the Kbase24. The annotated bins from NCBI were uploaded as GenBank file and a genome set was prepared using the app Batch Create Genome Set v1.2.0 along with one genome from the database (as default parameter was to take minimum one reference genome). The tree was computed by the alignment of a pre-decided subset of COG (Clusters of Orthologous Groups) domains using FastTree v. 2.1.1025, by maximum likelihood phylogeny. The tree was further annotated by iTOL v5.026. The reference genome was hidden during visualization, keeping only the MAGs under the study.
Data Records
The raw metagenomics reads and their corresponding pooled assemblies are available from EBI and NCBI27,28, respectively as detailed in Table 1. The sample-wise metagenome assemblies and pooled assemblies (GOC-A and GOK) are available under the Bioproject Id as mentioned in Table 129,30. The 309 assembled genome sequences and their functional annotations are available from NCBI database31,32 via biosample and genome accession numbers as detailed in Online-only Tables 1 and 2. The tree files corresponding to the figures and with reference genomes can be accessed throughfigshare33.
Technical Validation
The quality of MAGs was assessed using CheckM to validate the completeness and contamination of the bins. The genomes were also manually assessed at each point for similar bins by considering the parameters like GC, genome statistics and the number of genes.
Change history
17 September 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41597-021-01027-1
References
Moran, M. A. The global ocean microbiome. Science https://doi.org/10.1126/science.aac8455 (2015).
Tully, B. J., Graham, E. D. & Heidelberg, J. F. The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data https://doi.org/10.1038/sdata.2017.203 (2018).
Papudeshi, B. et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics https://doi.org/10.1186/s12864-017-4294-1 (2017).
Haroon, M. F., Thompson, L. R., Parks, D. H., Hugenholtz, P. & Stingl, U. A catalogue of 136 microbial draft genomes from Red Sea metagenomes. Sci. Data https://doi.org/10.1038/sdata.2016.50 (2016).
Sedlar, K., Kupkova, K. & Provaznik, I. Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Computational and Structural Biotechnology Journal https://doi.org/10.1016/j.csbj.2016.11.005 (2017).
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics https://doi.org/10.1093/bioinformatics/btv638 (2016).
Mootapally, C. et al. Antibiotic Resistome Biomarkers associated to the Pelagic Sediments of the Gulfs of Kathiawar Peninsula and Arabian Sea. Sci. Rep. https://doi.org/10.1038/s41598-019-53832-9 (2019).
Nathani, N. M., Mootapally, C. & Dave, B. P. Antibiotic resistance genes allied to the pelagic sediment microbiome in the Gulf of Khambhat and Arabian Sea. Sci. Total Environ. https://doi.org/10.1016/j.scitotenv.2018.10.409 (2019).
Bowers, R. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. https://doi.org/10.1038/nbt.3893 (2017).
Brochier-Armanet, C., Gribaldo, S. & Forterre, P. Spotlight on the Thaumarchaeota. ISME Journal https://doi.org/10.1038/ismej.2011.145 (2012).
Adam, P. S., Borrel, G., Brochier-Armanet, C. & Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME Journal https://doi.org/10.1038/ismej.2017.122 (2017).
Dyksma, S., Lenk, S., Sawicka, J. E. & Mußmann, M. Uncultured Gammaproteobacteria and Desulfobacteraceae Account for Major Acetate Assimilation in a Coastal Marine Sediment. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.03124 (2018).
Yilmaz, P., Yarza, P., Rapp, J. Z. & Glöckner, F. O. Expanding the world of marine bacterial and archaeal clades. Front. Microbiol. https://doi.org/10.3389/fmicb.2015.01524 (2016).
Hug, L. A. et al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ. Microbiol. https://doi.org/10.1111/1462-2920.12930 (2016).
Rasigraf, O., Schmitt, J., Jetten, M. S. M. & Lüke, C. Metagenomic potential for and diversity of N-cycle driving microorganisms in the Bothnian Sea sediment. Microbiologyopen https://doi.org/10.1002/mbo3.475 (2017).
Ngugi, D. K., Blom, J., Stepanauskas, R. & Stingl, U. Diversification and niche adaptations of Nitrospina-like bacteria in the polyextreme interfaces of Red Sea brines. ISME J. https://doi.org/10.1038/ismej.2015.214 (2016).
Spieck, E., Keuter, S., Wenzel, T., Bock, E. & Ludwig, W. Characterization of a new marine nitrite oxidizing bacterium, Nitrospina watsonii sp. nov., a member of the newly proposed phylum ‘Nitrospinae’. Syst. Appl. Microbiol. https://doi.org/10.1016/j.syapm.2013.12.005 (2014).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. https://doi.org/10.1101/gr.186072.114 (2015).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics https://doi.org/10.1093/bioinformatics/btz848 (2020).
Aziz, R. K. et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genomics https://doi.org/10.1186/1471-2164-9-75 (2008).
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature https://doi.org/10.1038/s41586-019-1058-x (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods https://doi.org/10.1038/nmeth.1923 (2012).
GraphPad Software, La Jolla California USA https://www.graphpad.com/ (2020).
Arkin, A. P. et al. KBase: The United States department of energy systems biology knowledgebase. Nature Biotechnology https://doi.org/10.1038/nbt.4163 (2018).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One https://doi.org/10.1371/journal.pone.0009490 (2010).
Letunic, I. & Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz239 (2019).
European Nucleotide Archive https://identifiers.org/ena.embl:ERP108616 (2020).
European Nucleotide Archive https://identifiers.org/ena.embl:ERP108617 (2020).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA623070 (2020).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA622989 (2020).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA598413 (2020).
NCBI Bioproject https://identifiers.org/ncbi/bioproject:PRJNA598416 (2020).
Nathani, N. M. et al. 309 metagenome assembled microbial genomes from deep sediment samples in the Gulfs of Kathiawar Peninsula. figshare https://doi.org/10.6084/m9.figshare.c.4944003 (2021).
Dagan, T., Roettger, M., Bryant, D. & Martin, W. Genome networks root the tree of life between prokaryotic domains. Genome Biol. Evol. https://doi.org/10.1093/gbe/evq025 (2010).
Acknowledgements
We acknowledge the boat crew and Dr. Paresh Poriya, Dr. Imtiyaz Belim and Dr. Jignesh Dabhi for their on board support in the sampling from the Gulfs. The work was funded by the Science and Engineering Research Board, Government of India, for support in the form of Early Career Research Award as National Post-Doctoral Fellowship to MCS and NMN under Grant No. PDF/2016/001239 and Grant No. PDF/2016/000190, respectively. We are extremely grateful to Prof. I. R. Gadhvi and Prof. Bharti P. Dave (Rtd.), Maharaja Krishnakumarsinhji Bhavnagar University for providing the administrative platform for execution of the projects. We are thankful to Prof. C. G. Joshi, Gujarat Biotechnology Research Centre for formal inputs in the data analysis.
Author information
Authors and Affiliations
Contributions
N.M.N.: planned the study, extracted DNA, assembled the metagenomes, binned and refined genomes, submitted all sequences to databases, and wrote the manuscript. K.J.D.: prepared table and annotated the genomes. P.V.: helped in writing the manuscript and genome quality assessment. M.S.M.: helped in submission of all sequences to databases. P.S.: helped in SNP density computation for all the bins. C.M.: planned the study, organized the sampling, collected the samples, extracted DNA, assembled the metagenomes, binned and refined the genomes, made figures, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Online-only Tables
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Nathani, N.M., Dave, K.J., Vatsa, P.P. et al. 309 metagenome assembled microbial genomes from deep sediment samples in the Gulfs of Kathiawar Peninsula. Sci Data 8, 194 (2021). https://doi.org/10.1038/s41597-021-00957-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00957-0
This article is cited by
-
Trait biases in microbial reference genomes
Scientific Data (2023)
-
Microbial community structure and exploration of bioremediation enzymes: functional metagenomics insight into Arabian Sea sediments
Molecular Genetics and Genomics (2023)
