
Abstract
Detailed descriptions of microbial communities have lagged far behind physical and chemical measurements in the marine environment. Here, we present 971 globally distributed surface ocean metagenomes collected at high spatio-temporal resolution. Our low-cost metagenomic sequencing protocol produced 3.65 terabases of data, where the median number of base pairs per sample was 3.41 billion. The median distance between sampling stations was 26 km. The metagenomic libraries described here were collected as a part of a biological initiative for the Global Ocean Ship-based Hydrographic Investigations Program, or “Bio-GO-SHIP.” One of the primary aims of GO-SHIP is to produce high spatial and vertical resolution measurements of key state variables to directly quantify climate change impacts on ocean environments. By similarly collecting marine metagenomes at high spatiotemporal resolution, we expect that this dataset will help answer questions about the link between microbial communities and biogeochemical fluxes in a changing ocean.
Measurement(s) | DNA sequencing • temperature of water • concentration of phosphate in water • concentration of nitrogen atom in water |
Technology Type(s) | Illumina sequencing • watercraft • continuous flow autoanalyzer |
Sample Characteristic - Organism | marine metagenome |
Sample Characteristic - Environment | ocean |
Sample Characteristic - Location | global |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13971122
Similar content being viewed by others

Background & Summary
A growing list of coordinated scientific efforts have produced deep metagenomic libraries of the surface ocean. Projects such as the Global Ocean Survey, Tara Oceans, and bioGEOTRACES1,2,3 have significantly advanced our understanding of marine microbial biogeography and biodiversity. However, this ever-increasing abundance of metagenomic data raises the question of how do we move beyond analyses of biodiversity to linking microbial traits with ecosystem function and elemental fluxes4. In oceanography, it has been widely acknowledged that sparse sampling results in high noise and error rates that in turn prevent the characterization of dynamic chemical balances and limit biogeochemical models5. Thus, we propose that an increased emphasis on high resolution spatio-temporal sampling of marine microbial communities would allow for a more mechanistic understanding of the relationship between microbes and ocean biogeochemistry.
The Global Ocean Ship-based Hydrographic Investigations Program (GO-SHIP) seeks to produce high spatial and vertical resolution measurements of physical, chemical, and biological parameters over the full water column. This internationally-organized program coordinates a network of sustained hydrographic sections that are repeatedly measured on an approximately decadal time scale. Compared to autonomous programs such as Argo, which has significantly increased the spatial and temporal resolution of ocean observations6, ship-based programs have the advantage of a much broader range of biogeochemical measurement capabilities and full water column coverage. To date, repeat hydrography programs have largely focused on physical (light, currents, water column thermohaline structure, etc.) and chemical (nutrients, oxygen, dissolved organic and inorganic carbon, pH, etc.) state variables. This work has significantly improved our understanding of the response of oxygen7, pH8, calcium carbonate saturation depth9, and sea level rise10 to global warming and anthropogenic carbon accumulation11. By comparison, systematic and sustained biological measurements of the microbial component of ocean ecosystems has lagged far behind.
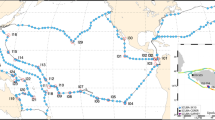
We present a dataset of 971 ocean surface water metagenomes collected at high spatio-temporal resolution in an effort to more mechanistically link marine microbial traits and biodiversity to both chemical and hydrodynamic ecosystem fluxes as a part of a novel Bio-GO-SHIP sampling program. Samples were collected in the Atlantic, Pacific, and Indian Ocean basins (Fig. 1, Table 1). This effort has been supported by GO-SHIP, SOCCOM, the Plymouth Marine Laboratory Atlantic Meridional Transect (PML AMT), and three National Science Foundation (NSF) Dimensions of Biodiversity funded cruises (AE1319, BVAL46, and NH1418) (Table 2). Whereas the median distance between Tara Oceans sampling stations was 709 km and the median distance between bioGEOTRACES sampling stations was 191 km, the median distance between sampling stations in the current Bio-GO-SHIP dataset is 26.5 km (Fig. 2). In addition, the majority of Bio-GO-SHIP samples were collected every 4–6 hours, allowing for analysis of diel fluctuations in microbial composition and gene content12. We anticipate that our high-resolution sampling scheme will allow for a more detailed examination of the relationship between the broad range of geochemical parameters measured across the various cruises (Table 2) and microbial diversity and traits.

Distribution of global surface microbial metagenomes from Bio-GO-SHIP (circles) in comparison to Tara Oceans (squares) and bioGEOTRACES (ovals). Symbol colours match the corresponding cruise name label colour.

Comparison of the distance between stations, station latitudes, and station longitudes for global surface ocean metagenomes. Individual station locations from (A) Bio-GO-SHIP, (B) bioGEOTRACES and (C) Tara Oceans were examined. Plots are labelled with the median value, M. Station distance was calculated as the distance to the nearest station.
Due to their rapid generation times and high diversity, microbial genomes integrate the impact of environmental change13 and can be used as a ‘biosensor’ of subtle biogeochemical regimes that cannot be identified from physical parameters alone12,14,15,16. Thus, the fields of microbial ecology and oceanography would benefit from coordinated, high resolution measurements of marine ‘omics products (i.e., metagenomes, metatranscriptomes, metaproteomes, etc.). This dataset provides an important example of the benefits of a high spatial and temporal resolution sampling regime. In addition, our data highlights the need for increased sampling of marine metagenomes in the Central and Western Pacific Ocean (Fig. 1), areas above 50°N and 50°S (Fig. 2), and below the euphotic zone. We hope and expect that these challenges will be addressed by the scientific community in the coming decade.
Methods
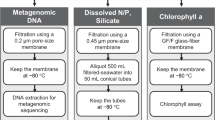
On all cruises, whole (i.e., no size fractionation) surface water was collected via either the Niskin rosette system (depth ~3–5 m) or the ship’s circulating seawater system (depth ~7 m). Between 2–10 L of surface water (Table 1) was collected in triple-rinsed containers and gently filtered through a 0.22 μm pore size Sterivex filter (Millipore, Darmstadt, Germany) using sterilized tubing and a Masterflex peristaltic pump (Cole-Parmer, Vernon Hills, IL). DNA was preserved with 1620 μL of lysis buffer (4 mM NaCl, 750 μM sucrose, 50 mM Tris-HCl, 20 mM EDTA) and stored at −20 °C before extraction.
To extract DNA (modified from Bostrom et al. 2004)17 Sterivex filters were incubated with 180 μL lysozyme (3.5 nM) at 37 °C for 30 minutes followed by an overnight 55 °C incubation with 180 μL Proteinase K (0.35 nM) and 100 μL 10% SDS buffer. DNA was extracted from the Sterivex with 1000 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA), precipitated in an ice-cold solution of 500 μL isopropanol (100%) and 1980 μL sodium acetate (3 mM, pH 5.2), pelleted via centrifuge for 30 mins at 4 °C, and resuspended in TE buffer in a 37°C water bath for 30 min. Next, DNA was purified using a genomic DNA Clean and Concentrator kit (Zymo Research Corp., Irvine, CA). Finally, DNA concentrations were quantified using a Qubit dsDNA HS Assay kit and Qubit fluorometer (ThermoFisher, Waltham, MA).
A total of 971 metagenomic libraries from 932 locations were prepared using Illumina-specific Nextera DNA transposase adapters and a Tagment DNA Enzyme and Buffer Kit (Illumina, San Diego, CA, cat. no. 20034197) (modified from Baym et al. 2015)18,19,20. Nextera adapter sequences to be used for bioinformatic quality trimming are: 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG-3′ and 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA G-3′. Custom Nextera DNA-style 8 bp unique dual index (UDI) barcodes I7 (5′-CAA GCA GAA GAC GGC ATA CGA GAT [NNN NNN NN]G TCT CGT GGG CTC GG-3′) and I5 (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC AC[N NNN NNN N]TC GTC GGC AGC GTC-3′) were used to multiplex the metagenomic libraries. A total of 1 μL of 2 ng μL−1 DNA was added to 1.5 μL tagmentation reactions (1.25 μL TD buffer, 0.25 μL TDE1) and incubated at 55 °C for 10 minutes. After tagmentation, product (2.5 μL) was immediately added to 22 μL reactions (1.02 μM per UDI barcode, 204 μM dNTPs, 0.0204 U Phusion High Fidelity DNA polymerase and 1.02X Phusion HF Buffer [ThermoFisher, Waltham, MA] final concentration). Barcodes were annealed to tagmented products using the following polymerase chain reaction (PCR): 72 °C for 2 min., 98 °C for 30 s., followed by 13 cycles of 98 °C 10 s., 63 °C 30 s., 72 °C 30 s., and a final extension step of 72 °C for 5 min.
To quality control tagmentation products, dimers that were less than 150 nucleotides long were removed using a buffered solution (1 M NaCl, 1 mM EDTA, 10 mM Tris-HCl, 44.4 M PEG-8000, 0.055% Tween-20 final concentration) of Sera-mag SpeedBeads (ThermoFisher, Waltham, MA). Metagenomic libraries were quantified using a Qubit dsDNA HS Assay kit (ThermoFisher, Waltham, MA) and a Synergy 2 Microplate Reader (BioTek, Winooski, VT). Libraries were then pooled at equimolar concentrations. Pooled library concentration was verified using a KAPA qPCR platform (Roche, Basel, Switzerland). Finally, dimer removal as well as read size distribution were checked using a 2100 Bioanalyzer high sensitivity DNA trace (Agilent, Santa Clara, CA).
54 samples were sequenced on two Illumina HiSeq 4000 lanes using 150 bp paired-end chemistry with 300 cycles (Illumina, San Diego, CA). A total of 666 samples were sequenced on three Illumina NovaSeq S4 flowcells and an additional 251 samples were sequenced on a combination of S1 and SP flowcells using 150 bp paired-end chemistry with 300 cycles. The sequencing strategy produced a total of 2.42 × 1010 reads, or 3.65 × 1012 bp. The median number of bases per sample was 3.41 billion (range: 61,400,000–21.4 billion). Prior to read trimming and quality filtering, 74% of all forward and reverse reads had an average quality score ≥Q25 (Table 1). The sequencing cost per bp in US dollars was $8.2 × 10−9.
Data Records
The majority of the samples here were collected under the auspices of the international GO-SHIP program and the national programs that contribute to it21,22,23,24. Links to publicly available metadata variables collected via CTD cast are provided in Table 2. A comprehensive data directory of all metadata resources, including those that were collected and may be requested from individual PIs, is available through GO-SHIP and the Carbon and Climate Hydrographic Data Office (CCDHO) under a Public Domain Mark (PDM).
Metadata variables from the AMT-28 cruise are hosted by the British Oceanographic Data Centre (BODC)25 and the Southern Ocean Carbon and Climate Observations and Modeling project (SOCCOM).
The BVAL46, AE1319, and NH1418 cruises were collected as a part of the “Biological Controls on the Ocean C:N:P Ratios” project funded by the NSF Division of Ocean Sciences26,27,28,29. Data associated with these deployments are hosted by the NSF Biological and Chemical Oceanography Data Management Office (BCO-DMO) under Project 2178 and are archived by the Woods Hole Open Access Server (WHOAS) under a Creative Commons BY 4.0 (CC BY 4.0) license.
All sequencing products associated with the Bio-GO-SHIP program can be found under BioProject ID PRJNA656268 hosted by the National Center for Biotechnology Information Sequence Read Archive (SRA)30. SRA accession numbers associated with each metagenome file are provided in Supplementary Table 1.
Technical Validation
To ensure that no contamination of metagenomes occurred, negative controls were used. To ensure optimum paired-end short read sequencing, a 2100 Bioanalyzer high sensitivity DNA trace (Agilent, Santa Clara, CA) was used for each library to confirm that ~90% of the sequence fragments were above 200 bp and below 600 bp in length (Table 3). A Qubit (ThermoFisher, Waltham, MA) and a KAPA qPCR platform (Roche, Basel, Switzerland) were used to ensure that all pooled libraries were submitted for sequencing at a concentration > 15 nM.
Usage Notes
The genomic data described here have not been pre-screened or processed in any way. We recommend quality control parameters. Prior to our sequence analysis in subsequent projects, we removed adapter sequences, performed sequence quality control, and ensured there was no contamination from common genomic add-ins such as Phi-X using the following code parameters:
Trimmomatic (v0.35): PE ILLUMINACLIP:NexteraPE-PE.fa:2:30:10 SLIDINGWINDOW:4:15 MINLEN:36
BBMap (v37.50): bbduk.sh -Xmx1g ref = /BBMap/37.50/resources/phix174_ill.ref.fa.gz k = 31 hdist = 1
Nutrient data (NO3, NO2, PO4, SiO4) collected by SOCCOM and funded by the National Science Foundation are available from the AMT-28 transect through the CCHDO (http://cchdo.ucsd.edu, search on SOCCOM).
Code availability
Custom scripts were not used to generate or process this dataset. Software versions and non-default parameters used have been appropriately specified where required.
References
Rusch, D. B. et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. 5, e77 (2007).
Pesant, S. et al. Open science resources for the discovery and analysis of Tara Oceans data. Sci. Data 2, 150023 (2015).
Biller, S. J. et al. Marine microbial metagenomes sampled across space and time. Sci. Data 6, 180176 (2019).
Hall, E. K. et al. Understanding how microbiomes influence the systems they inhabit. Nat. Microbiol. 3, 977–982 (2018).
Curtin, T. B., Bellingham, J. G., Catipovic, J. & Webb, D. Autonomous oceanographic sampling networks. Oceanography 6, 86–94 (1993).
Riser, S. C. et al. Fifteen years of ocean observations with the global Argo array. Nat. Clim. Change 6, 145–153 (2016).
Schmidtko, S., Stramma, L. & Visbeck, M. Decline in global oceanic oxygen content during the past five decades. Nature 542, 335–339 (2017).
Doney, S. C., Fabry, V. J., Feely, R. A. & Kleypas, J. A. Ocean acidification: The other CO2 problem. Ann. Rev. of Mar. Sci. 1, 169–192 (2009).
Feely, R. A. et al. Impact of anthropogenic CO2 on the CaCO3 system in the oceans. Science 305, 362–366 (2004).
Purkey, S. G. & Johnson, G. C. Warming of global abyssal and deep Southern Ocean waters between the 1990s and 2000s: Contributions to global heat and sea level rise budgets. J. Clim. 23, 6336–6351 (2010).
Gruber, N. et al. The oceanic sink for anthropogenic CO2 from 1994 to 2007. Science 363, 1193–1199 (2019).
Larkin, A. A. et al. Subtle biogeochemical regimes in the Indian Ocean revealed by spatial and diel frequency of Prochlorococcus haplotypes. Limnol. Oceanogr. 65, S220–S232 (2020).
Larkin, A. A. & Martiny, A. C. Microdiversity shapes the traits, niche space, and biogeography of microbial taxa. Environ. Microbiol. Rep. 9, 55–70 (2017).
Garcia, C. A. et al. Linking regional shifts in microbial genome adaptation with surface ocean biogeochemistry. Philos. Trans. Roy. Soc. Lond. B. 375, 20190254 (2020).
Martiny, A. C., Ustick, L., Garcia, C. A. & Lomas, M. W. Genomic adaptation of marine phytoplankton populations regulates phosphate uptake. Limnol. Oceanogr. 65, S340–S350 (2020).
Sonnewald, M., Dutkiewicz, S., Hill, C. & Forget, G. Elucidating ecological complexity: Unsupervised learning determines global marine eco-provinces. Sci. Adv. 6, eaay4740 (2020).
Bostrom, K. H., Simu, K., Hagstrom, A. & Riemann, L. Optimization of DNA extraction for quantitative marine bacterioplankton community analysis. Limnology and Oceanography: Methods 2, 365–373 (2004).
Baym, M. et al. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10, e0128036 (2015).
Wandro, S. et al. Predictable molecular adaptation of coevolving Enterococcus faecium and lytic phage EfV12-phi1. Front. Microbiol. 9, 3192 (2019).
Oliver, A. et al. Cervicovaginal microbiome composition is associated with metabolic profiles in healthy pregnancy. mBio 11, e01851–20 (2020).
Barbero, L. Bottle data from Cruise 33RR20160321, exchange version. CCHDO: CLIVAR and Carbon Hydrographic Data Office https://doi.org/10.7942/C2008W (2016).
Sonnerup, R., Carter, B., Purkey, S. & Bourbonnais, A. Bottle data from Cruise 33RO20161119, exchange version. CCHDO: CLIVAR and Carbon Hydrographic Data Office https://doi.org/10.7942/C21T0F (2016).
Volkov, D. & Menezes, V. Bottle data from Cruise 33RO20180423, exchange version. CCHDO: CLIVAR and Carbon Hydrographic Data Office https://doi.org/10.7942/C25H2B (2018).
Barbero, L. & Schatzman, C. Bottle data from Cruise 33RO20200321, exchange version. CCHDO: CLIVAR and Carbon Hydrographic Data Office https://doi.org/10.7942/C2894Z (2020).
Bargery, A. AMT28 (JR18001) CTD profiles (pressure, temperature, salinity, potential temperature, density, fluorescence, transmittance, downwelling PAR, dissolved oxygen concentration) calibrated and binned to 1 dbar. British Oceanographic Data Centre, National Oceanography Centre, NERC, UK https://doi.org/10.5285/b8c6056a-db74-7453-e053-6c86abc00ce5 (2021).
Lomas, M. W., Martiny, A. C., Bonachela, J. A. & Levin, S. Biogeochemistry of microbial phosphorus uptake from cruises in the Sargasso Sea Bermuda Atlantic Time-Series Station from 2011-2013 (Biological C:N:P ratios project). Biological and Chemical Oceanography Data Management Office (BCO-DMO) https://doi.org/10.26008/1912/bco-dmo.538091.2 (2014).
Lomas, M. W. & Martiny, A. Depth profile data from Bermuda Atlantic Time-Series Validation cruise 46 (BVAL46) in the Sargasso Sea from Sept-Oct. 2011. Biological and Chemical Oceanography Data Management Office (BCO-DMO) https://doi.org/10.26008/1912/bco-dmo.829843.1 (2020).
Lomas, M. W. & Martiny, A. Depth profile data from R/V Atlantic Explorer AE1319 in the NW Atlantic from Aug-Sept. 2013. Biological and Chemical Oceanography Data Management Office (BCO-DMO) https://doi.org/10.26008/1912/bco-dmo.829797.1 (2020).
Lomas, M. W. & Martiny, A. Depth profile data from R/V New Horizons New Horizons NH1418 in the tropical Pacific from Sept-Oct. 2014. Biological and Chemical Oceanography Data Management Office (BCO-DMO) https://doi.org/10.26008/1912/bco-dmo.829895.1 (2020).
NCBI Sequence Read Archive: http://identifiers.org/ncbi/insdc.sra:SRP278138 (2020).
Acknowledgements
We would like to thank the captains and crew of the R/V Atlantic Explorer, R/V New Horizon, NOAA Ship Ronald H. Brown, R/V Roger Revelle, and the R.R.S. James Clark Ross. Our additional thanks go to Michael Lomas (Bigelow Laboratory for Ocean Sciences) and Andy Rees (Plymouth Marine Laboratory). Financial support for this project was provided by the National Science Foundation (OCE-1046297, 1559002, 1848576, and 1948842 to ACM). LJU was supported by the National Institutes of Health (T32AI141346). LB and DLV were supported in part under the auspices of the Cooperative Institute for Marine and Atmospheric Studies (CIMAS), a cooperative institute of the University of Miami and NOAA (cooperative agreement NA10OAR4320143). The PML AMT is funded by the UK Natural Environment Research Council through its National Capability Long-term Single Centre Science Programme, Climate Linked Atlantic Sector Science (grant number NE/R015953/1). This study contributes to the international IMBeR project and is contribution number 357 of the AMT program. SOCCOM nutrient sampling (LDT) on AMT-28 was supported by the National Science Foundation (OCE-PLR-1425989). GO-SHIP is supported in part by the NOAA Global Ocean Monitoring and Observation program (U8R1SE3-PRF) and by the National Science Foundation (OCE-1437015). P18, I07N, and A13.5 (aborted) were NOAA-led cruises and I09N was an NSF-led cruise.
Author information
Authors and Affiliations
Contributions
A.A.L. wrote the manuscript, designed metagenomic protocols, coordinated sample collection, collected/processed samples, performed and/or supervised all metagenomic sequencing, and compiled metadata. C.A.G. coordinated sample collection, collected/processed samples, performed metagenomic sequencing, and compiled metadata. N.G. coordinated sample collection, collected/processed samples, and performed metagenomic sequencing. M.L.B. performed metagenomic sequencing and compiled metadata. J.A.L. collected/processed samples and performed metagenomic sequencing. L.J.U. processed samples and compiled metadata. L.B., B.G.C., R.E.S., L.D.T. and D.L.V. coordinated GO-SHIP and SOCCOM (L.D.T.) collection and collaboration efforts. G.T. coordinated PML AMT/GO-SHIP collection and collaboration efforts. A.C.M. designed and supervised the study, secured funding, and coordinated GO-SHIP collection. All authors contributed to manuscript editing and revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Larkin, A.A., Garcia, C.A., Garcia, N. et al. High spatial resolution global ocean metagenomes from Bio-GO-SHIP repeat hydrography transects. Sci Data 8, 107 (2021). https://doi.org/10.1038/s41597-021-00889-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00889-9
This article is cited by
-
Decoding populations in the ocean microbiome
Microbiome (2024)
-
Genome-scale community modelling reveals conserved metabolic cross-feedings in epipelagic bacterioplankton communities
Nature Communications (2024)
-
Marine picoplankton metagenomes and MAGs from eleven vertical profiles obtained by the Malaspina Expedition
Scientific Data (2024)
-
Basin-scale biogeography of Prochlorococcus and SAR11 ecotype replication
The ISME Journal (2023)
-
Global scale phylogeography of functional traits and microdiversity in Prochlorococcus
The ISME Journal (2023)
