
Abstract
Caligus rogercresseyi, commonly known as sea louse, is an ectoparasite copepod that impacts the salmon aquaculture in Chile, causing losses of hundreds of million dollars per year. In this study, we report a chromosome-scale assembly of the sea louse (C. rogercresseyi) genome based on single-molecule real-time sequencing (SMRT) and proximity ligation (Hi-C) analysis. Coding RNAs and non-coding RNAs, and specifically long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) were identified through whole transcriptome sequencing from different life stages. A total of 23,686 protein-coding genes and 12,558 non-coding RNAs were annotated. In addition, 6,308 lncRNAs and 5,774 miRNAs were found to be transcriptionally active from larvae to adult stages. Taken together, this genomic resource for C. rogercresseyi represents a valuable tool to develop sustainable control strategies in the salmon aquaculture industry.
Measurement(s) | sequence_assembly • RNA • genome • DNA |
Technology Type(s) | sequence assembly process • RNA sequencing • DNA sequencing |
Factor Type(s) | developmental stage • sex |
Sample Characteristic - Organism | Caligus rogercresseyi |
Sample Characteristic - Environment | aquaculture process |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13186865
Similar content being viewed by others

Background & Summary
Sea lice are marine copepods that negatively impact the salmon aquaculture worldwide. Two of the most studied sea lice species are Caligus rogercresseyi and Lepeophtheirus salmonis1,2. Annually the salmon farming industry accounts $480 million in losses associated with sea lice, representing 10% of production costs3,4,5. The parasitism on farmed fish causes skin damage, immunosuppression, and co-infection of opportunistic pathogenic bacteria6,7,8. Like all ectoparasites, lice spend a large part of their life cycle on a fish host, displaying specific mechanisms for evading the host’s immune response9,10,11.
The life cycle of lice species is complex and consists of several instars divided by moults. For instance, C. rogercresseyi comprises two larval stages (nauplius I, nauplius II and copepodite), four juvenile stages (chalimus I - IV) and one adult stage (female or male)12. During the copepodite stage, the process of host identification occurs, preparing the lice for infestation and settlement8. The successful infestation process on the host allows the parasite access to nutrients for reproduction and adult development13,14. Previous studies have shown that lice have developed physical mechanisms of host recognition. Among these, lice can identify the temperature of the water, salinity changes, and detect the swimming of fish15. Host identification via detection of semiochemicals has also been reported16. In C. rogercresseyi, the presence of advanced chemoreceptors that are capable of identifying specific molecules of different host species has recently been described17,18,19. Herein, the gene family of ionotropic receptors (IRs) are pivotal molecular components for the salmon-louse interaction20,21.
Molecular understanding of C. rogercresseyi is pivotal to develop sustainable salmon aquaculture. However, genomic resources in this species are limited and poorly characterized at functional levels. In 2012, Yasuike et al. (2012) reported a compilation of genomic information on different sea lice genera, including C. rogercresseyi. It was not until 2014 that Gallardo-Escárate et al.22 reported the transcriptome of different life stages during the ontogenetic development as well as differences between male and female adults. This transcriptomic resource served as a basis to identify genes involved in molting, cuticle formation, myogenesis, metabolism, immune response, nervous system development and reproduction. Notably, this gene set has served as a basis for the design of new vaccines2.
The increasing availability of transcriptome data has revealed the importance of non-coding RNAs as key regulators of the mRNA transcription23. To date, microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have been studied in several arthropod species with special emphasis on parasitic vectors24. Long non-coding RNAs are sequences greater than 200 nucleotides, transcribed in a similar way as coding RNAs25. It has been suggested that the number of lncRNAs has increased during evolution, where organisms with more complex mechanisms have acquired more lncRNAs to control diversifying biological processes. MicroRNAs are transcripts around 22 base pairs in length that play an important role in post-transcriptional gene regulation26. There are studies that show that miRNAs are not only regulators of biological processes, but can also participate in parasite-host interaction processes9,27. In insects affected by viruses, it has been observed that viruses are capable of releasing miRNAs that can regulate the expression of their host genes in order to successfully establish the infection28. For C. rogercresseyi, several miRNAs expressed during the different stages of development have been characterized29,30. Within the profile of miRNAs characterized in C. rogercresseyi, the miRNA annotated as Bantam is highly expressed in the infective stage of copepodid. This suggests that Bantam has a key role in the success of the infection. Taken together, these resources reported for the sea louse C. rogercresseyi represent a valuable tool to develop sustainable control strategies in the salmon industry. What is lacking is an annotated genome that will facilitate an integrated examination of molecular interactions and provide insight in evolutionary and epigenetics processes that underlie critical life history characteristics. In this study, we report the chromosome-scale whole genome sequence of C. rogercresseyi through application of Pacific Biosciences’ single molecule sequencing technique (SMRT) and Phase Genomics’ proximity ligation (Hi-C) based genome scaffolding.
Methods
Sample collection, NGS libraries, and sequencing
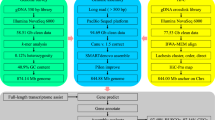
Adult female specimens of Caligus rogercresseyi were collected from Atlantic salmon (Salmo salar) at the Caligus Reference Laboratory (CRL), University of Concepción, Chile (Fig. 1). With the aims to reduce the heterozygosity or the number of individuals per pool, female lice were selected for whole-genome sequencing. The samples were frozen in liquid nitrogen to preserve DNA quality, and ten females were used for genomic DNA isolation. High quality DNA was isolated using the Qiagen DNA purification kit (QIAGEN, Germantown, MD, USA) following the manufacturer’s instructions. It is important to note that sea lice are marine copepods exposed to marine environmental conditions and consequently to commensal microorganisms. To reduce bacterial DNA contamination, lice were treated with 20 mg/ml ampicillin (Sigma-Aldrich, USA), 20 mg/ml Kanamycin (US biological, USA), 1x Penicillin-Streptomycin (GIBCO, USA), 100 ug/ml Primocin (Invivogen, USA) for 72 hr prior to the DNA extraction protocol31. Furthermore, lice from different developmental stages were separately collected, fixed in RNA Later solution (Ambion, USA), and stored at −80 °C until RNA extractions.

The sea louse Caligus rogercresseyi. Adult female (right) and adult male (left). Magnification 10x.
Genomic DNA libraries were constructed according to the manufacturer’s protocols for genome assembly (Table 1). SMRT sequencing yielded 38.32 Gb long reads from 8 SMRT cells (Table 1S). The subreads N50 and average lengths were 11,093 and 6,824 bp, respectively. Hi-C libraries were constructed from whole animals using Phase Genomics’ Animal Hi-C kit and sequenced on an Illumina’s Hiseq4000 platform to yield 238 million of reads. Short-read sequencing libraries were prepared using an insert size of 150 bp obtained from 1 μg of genomic DNA, after fragmentation, end-paired, and ligation to adaptors, respectively. The ligated fragments were fractionated on agarose gels and purified by PCR amplification to produce sequencing libraries.
For transcriptome sequencing, RNA libraries were constructed from nauplius I, nauplius II, copepodid, Chalimus I-II, Chalimus III-IV, males and females, and sequenced by Illumina technology according to the manufacturer’s protocols (Table 1). Briefly, total RNA was extracted from 10 parasites from each stage using the Trizol reagent method (Invitrogen, USA). The quality and integrity of extracted RNAs was measured in a TapeStation 2200 instrument (Agilent, USA), using the R6K Reagent Kit based on manufacturer’s instructions. RNA samples >9 in RIN numbers were selected for library preparation. For whole transcriptome sequencing, 2 μg of total RNA was used for dscDNA libraries with TruSeq Total RNA kit (Illumina, USA). RNA libraries quantification was conducted by qPCR using the NEBNext Library Quant Kit for Illumina (New England Biolabs, USA). The sequencing was performed using the MiSeq platform (Illumina, USA) using a 2 × 250 bp paired-end reads scheme (single flow cell per developmental stage). In addition to generating conventional RNA-seq for 6 developmental stages, small-RNA libraries were also constructed using TruSeq Small RNA Kit (Illumina, USA) for each stage, with libraries run in 41 single-end cycles. Small-RNA libraries were simultaneously sequenced using barcodes according to the manufacturer’s protocols. In total 3 flow cells were used to sequence the 6 developmental stages. A total of 52.01 and 28.18 Gb was yielded for transcriptome and miRNome characterization, respectively (Table 1).
De novo assembly of C. rogercresseyi genome
With eight single-molecular real-time cells in the PacBio Sequel platform, we generated 38.32 Gb of high-quality DNA genome information. These long subreads were assembled with the Canu V1.5 package32 using default parameters, yielding a draft genome for the sea louse equivalent to 727 Mb with contig N50 of 43,366 bp and 35.55 GC%. The draft genome was assembled with CANU in 25,608 contigs (Table 2). The size genome assembly made by CANU was comparable with previous genome size reported for closely related species33,34. However, the manual curation of a subset of contigs revealed bacterial DNA contamination. As we previously mentioned, antibiotic treatment was applied to reduce the natural lice microbiota. However, it appeared that some fraction of the bacterial burden still remained despite the antimicrobial compound used. To reduce the bacterial DNA contamination, all contigs assembled by Canu were firstly filtered against NCBI prokaryotic reference sequence database and then against the reference C. rogercresseyi transcriptome (Table 3). For the first filter, BLASTx was applied with an expectation value of 10.0, word size = 3, filter low complexity, protein matrix and gap costs = BLOSUM62, Existence, 11-1, meanwhile that for the second filter a mapping approach was implemented with the following settings using CLC Genomics Workbench V12 (Qiagen, USA): match score = 1, mismatch cost = 2, cost of insertions and deletions = Linear gap cost, insertion cost = 3, deletion cost = 3, length fraction = 0.5, similarity fraction = 0.8, global alignment = No, non-specific match handling = map randomly. Taking advantage of the two filters, we removed all the contigs with a significant match to bacterial DNA, reducing the number of contigs produced by the draft genome for C. rogercresseyi made by Canu from 25,608 to 17,711 contigs. Here, the new dataset yielded a draft genome assembly of 519.19 Mb with an N50 of 38,179 bp (Table 4). Notably, the DNA contamination produced by the natural microbiota found in C. rogercresseyi was ∼30%. This fact shows the importance of the microbiota in louse biology, revealing putative associations with the pathogenesis of this ectoparasite.
Chromosome assembly of C. rogercresseyi using chromatin interaction mapping analysis
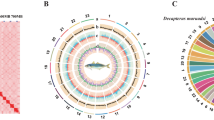
In vivo Hi-C is a technique that maps physical DNA-DNA proximity across the entire genome35,36. The method was introduced as a genome-wide version of its predecessor, 3 C (Chromosome Conformation Capture)37, and has been used as a powerful tool in chromosome-scale genome assembly of many animals in recent years. In this study, Hi-C experiments and data analysis on adult females were used for the chromosome assembly of the sea louse C. rogercresseyi. Here, two Hi-C libraries were prepared and sequenced by Phase Genomics (Seattle, WA, USA), resulting in ∼100x coverage and ∼238 million 150-bp paired-end reads (Table 4). The Hi-C analysis evidenced that 46.70% of high-quality reads analysed showed intercontig signals or Cis-close position (<10kbp on the same contig), and an additional 5.32% of sequence reads revealed a Cis-far conformation (>10Kbp on the same contig). To order and orient the 17,711 contigs Hi-C reads were aligned using Bowtie238 and scaffolding performed using Proximo (Phase Genomics, Seattle, WA, USA). We then applied Juicebox39 for visual inspection and manual correction. We also manually removed 7,897 scaffolds that were microbe-sized and disconnected from the rest of the assembly. We obtained the first chromosome-level high-quality C. rogercresseyi assembly with an N50 scaffold of 29.78 Mb, providing a useful genomic resource for research in sea louse biology and also, to develop novel control strategies applied to the salmon aquaculture (Table 5). In order to visualize the scaffold’s length construction, the in vivo Hi-C data were used to generate 21 pseudo-chromosomes assembled with PacBio consensus long DNA reads (Fig. 2). The largest scaffold was assembled from 1,235 contigs, a size of 36.77 Mb. Meanwhile, the smallest scaffold was 7.98 Mb of length and consisted of 396 original contigs (Fig. 3). Notably, the number of contigs in scaffolds were 16,931 (100% of all contigs in chromosome clusters, 95.6% of all contigs) and 505.27 Mb of genome size (100% of all length in chromosome cluster, 97.33% of all sequence length). The completeness of genome assembly was assessed by the single-copy ortholog set (BUSCO, V3.0.2) against Eukaryota, Metazoa, and Arthropoda40. The results indicated a complete BUSCO of 78.9% [S:75.3%, D:3.6%] and a fragmented BUSCO of 13.5% [M:13.6%, n:303].

The sea louse Caligus rogercresseyi genome contig contact matrix using Hi-C data. The blue squares represent the draft scaffold. The color bar illuminates the Hi-C contact density in the plot.

The sea louse Caligus rogercresseyi genome. The circos plot shows the genomic features for the 21 pseudo-chromosomes. A) GC content, B) Gene density, C) lncRNA density, D) miRNA density and E) Repetitive elements. The chromosome size is shown in Mb scale.
Repetitive element and non-coding gene annotation in the C. rogercresseyi genome
Repetitive elements and non-coding genes in the sea louse genome were annotated by homologous comparison and ab initio prediction. RepeatMasker41 was used for homologous comparison by searching against the Repbase database and RepeatModeler42. According to these analyses, about 269.83 Mb Mb of repeat sequences were annotated, which accounted for 51.9% of the whole genome. Herein, DNA transposons, LINE, and LTR transposable elements were identified (Table 6). Useful genome information for population genetic studies is the identification of simple sequence repeats (SSRs) or microsatellites. The mining of SSRs revealed that the C. rogercresseyi genome has 441,494 SSR sequences, where 65.76% represent dinucleotide motifs (Table 7). The total of SSR sequences accounted for 1.39% of the whole genome, and the genome distribution was correlated with the chromosome size (Fig. 1S). Furthermore, SSRs type dinucleotides, and specifically, the motifs AC/GT were the most abundant, representing the 65.76% of the total microsatellite sequences (Fig. 2S). Trinucleotides and tetranucleotides were found in 32.46% of the SSRs sequences (Fig. 3 and Table 7).
Protein-coding genes prediction and functional annotation in the C. rogercresseyi genome
For the identification of protein-coding genes, two approaches were employed for the sea louse genome, including homologous comparison and ab initio prediction. For homologous comparison, the protein sequences from Caenorhabditis elegans (GCA_000002985.3), Drosophila melanogaster (GCA_000001215.4), and Daphnia pulex (GL732539.1) genomes were extracted using the respectively published genomes. and aligned against the sea louse genome using TBLASTN (e-value < 1e-5). Gene sequence structure of each candidate genes was predicted using GeneWise43. For ad initio prediction, five tools were used to predict protein-coding genes using the Genome Sequence Annotation Server “GenSAS” (https://www.gensas.org)44. Specifically, Augustus, Braker, GeneMarkES, SNAP, and GlimmerM were used with default parameters. Finally, a non-redundant reference gene set was generated using EvidenceModeler (EVM) and PASA2 tools45. Taken together 25,510 protein-coding genes were identified. (Fig. 3 and Table 8). Additionally, 437 tRNAs were predicted using tRNAscan-SE, and 39 rRNA genes were annotated using RNAmmer via GenSAS. For non-coding RNAs with putative regulatory roles, 5,774 miRNAs and 6,308 long-ncRNAs were identified and annotated within the C. rogercresseyi genome using transcriptome sequencing data (Fig. 4 and Table 9). For functional annotation, the predicted proteins within the sea louse genome were searched by homology against four databases of InterPro46, GO47, KEGG KO48, and Swissprot49. Overall, 88.05%, 68.85%, 64.02%, and 91.02% of genes matched entries in these databases, respectively. A total of 23,686 genes (93%) were successfully annotated by gene function and conserved protein motifs (Table 10).

Stage-specific transcriptome analysis in the sea louse Caligus rogercresseyi. (A) Transcriptome patterns of coding genes during the lifecycle. The heatmap was based on Transcripts Per Million (TPM) calculation and hierarchical clustering on Manhattan distances with average linkage. White colors mean upregulated coding genes, blue colors downregulated genes. (B) Venn diagram showing shared and unique genes expressed among the six developmental stages. (C) GO enrichment of stage-specific genes (P-value ≤ 10–16;|fold-change| > 5) annotated for key biological processes differentially expressed. The radar plot represents the comparison between two developmental stages according the C. rogercresseyi lifecycle.
Technical Validation
RNA integrity
Before constructing RNA-seq libraries, the concentration and quality of total RNA were evaluated using Agilent 2100 Bioanalyser (Agilent, USA). Three metrics, including total amount, RNA integrity, and rRNA ratio, were used to estimate the content, quality, and degradation level of RNA samples. In this study, only total RNAs with a total amount of ≥10 μg, RNA integrity number ≥8, and rRNA ratio ≥1.5 were finally subjected to construct the sequencing library.
Quality filtering of Illumina sequencing raw reads
The initial raw sequencing reads were evaluated in terms of the average quality score at each position, GC content distribution, quality distribution, base composition, and other metrics. Furthermore, the sequencing reads with low quality were also filtered out before the genome assembly and annotation of gene structure.
Data Records
DNA and RNA sequencing runs were deposited to NCBI Sequence Read Archive (SRA)50,51,52. The assembled genome has been deposited at NCBI assembly with the accession number ASM1338718v153. Additional files containing repeated sequences, gene structure, and functional prediction were deposited in the Figshare database54.
Code availability
The sequence data were generates using the Genome Sequence Annotation Server “GenSAS” (https://www.gensas.org)44. No custom computer codes were generated in this work (Table 11).
References
Costelloe, M., Costelloe, J., O’Donohoe, G., Coghlan, N. & O’Connor, B. A review of field studies on the sea louse, Lepeophtheirus salmonis Kroyer on the west coast of Ireland. Bull Eur Assn Fish P 19, 260–264 (1999).
Gallardo-Escarate, C. et al. The race between host and sea lice in the Chilean salmon farming: a genomic approach. Rev Aquacult 11, 325–339, https://doi.org/10.1111/raq.12334 (2019).
Dresdner, J. et al. Impact of Caligus treatments on unit costs of heterogeneous salmon farms in Chile. Aquacult Econ Manag 23, 1–27, https://doi.org/10.1080/13657305.2018.1449271 (2019).
Costello, M. J. How sea lice from salmon farms may cause wild salmonid declines in Europe and North America and be a threat to fishes elsewhere. Proc. R. Soc. B-Biol. Sci. 276, 3385–3394, https://doi.org/10.1098/rspb.2009.0771 (2009).
Abolofia, J., Wilen, J. E. & Asche, F. The Cost of Lice: Quantifying the Impacts of Parasitic Sea Lice on Farmed Salmon. Mar Resour Econ 32, 329–349, https://doi.org/10.1086/691981 (2017).
Gallardo-Escarate, C. et al. Catching the complexity of salmon-louse interactions. Fish Shellfish Immunol 90, 199–209, https://doi.org/10.1016/j.fsi.2019.04.065 (2019).
Arriagada, G. et al. Caligus rogercresseyi infestation is associated with Piscirickettsia salmonis-attributed mortalities in farmed salmonids in Chile. Prev Vet Med 171, 104771, https://doi.org/10.1016/j.prevetmed.2019.104771 (2019).
Fast, M. D. Fish immune responses to parasitic copepod (namely sea lice) infection. Dev. Comp. Immunol. 43, 300–312, https://doi.org/10.1016/j.dci.2013.08.019 (2014).
Valenzuela-Munoz, V., Novoa, B., Figueras, A. & Gallardo-Escarate, C. Modulation of Atlantic salmon miRNome response to sea louse infestation. Dev Comp Immunol 76, 380–391, https://doi.org/10.1016/j.dci.2017.07.009 (2017).
Valenzuela-Munoz, V. & Gallardo-Escarate, C. Iron metabolism modulation in Atlantic salmon infested with the sea lice Lepeophtheirus salmonis and Caligus rogercresseyi: A matter of nutritional immunity? Fish Shellfish Immunol. 60, 97–102, https://doi.org/10.1016/j.fsi.2016.11.045 (2017).
Valenzuela-Munoz, V., Boltana, S. & Gallardo-Escarate, C. Uncovering iron regulation with species-specific transcriptome patterns in Atlantic and coho salmon during a Caligus rogercresseyi infestation. J Fish Dis 40, 1169–1184, https://doi.org/10.1111/jfd.12592 (2017).
Gonzalez, L. & Carvajal, J. Life cycle of Caligus rogercresseyi, (Copepoda: Caligidae) parasite of Chilean reared salmonids. Aquaculture 220, 101–117, https://doi.org/10.1016/S0044-8486(02)00512-4 (2003).
Goncalves, A. T., Farlora, R. & Gallardo-Escarate, C. Transcriptome survey of the lipid metabolic pathways involved in energy production and ecdysteroid synthesis in the salmon louse Caligus rogercresseyi (Crustacea: Copepoda). Comp Biochem Phys B 176, 9–17, https://doi.org/10.1016/j.cbpb.2014.07.002 (2014).
Farlora, R., Araya-Garay, J. & Gallardo-Escarate, C. Discovery of sex-related genes through high-throughput transcriptome sequencing from the salmon louse Caligus rogercresseyi. Mar Genomics 15, 85–93, https://doi.org/10.1016/j.margen.2014.02.005 (2014).
Mordue, A. J. & Birkett, M. A. A review of host finding behaviour in the parasitic sea louse, Lepeophtheirus salmonis (Caligidae: Copepoda). J. Fish Dis. 32, 3–13, https://doi.org/10.1111/j.1365-2761.2008.01004.x (2009).
Fields, D. M., Weissburg, M. J. & Browman, H. Chemoreception in the salmon louse Lepeophtheirus salmonis: an electrophysiology approach. Diseases of Aquatic Organisms 78, 161–168, https://doi.org/10.3354/dao01870 (2007).
Nunez-Acuna, G. et al. The Atlantic salmon (Salmo salar) antimicrobial peptide cathelicidin-2 is a molecular host-associated cue for the salmon louse (Lepeophtheirus salmonis). Sci Rep 8, 13738, https://doi.org/10.1038/s41598-018-31885-6 (2018).
Nunez-Acuna, G., Gallardo-Escarate, C., Skiftesvik, A. B., Fields, D. M. & Browman, H. I. Silencing of ionotropic receptor 25a decreases chemosensory activity in the salmon louse Lepeophtheirus salmonis during the infective stage. Gene 697, 35–39, https://doi.org/10.1016/j.gene.2019.02.012 (2019).
Ingvarsdottir, A. et al. Semiochemical strategies for sea louse control: host location cues. Pest Manag Sci 58, 537–545, https://doi.org/10.1002/ps.510 (2002).
Nunez-Acuna, G., Valenzuela-Munoz, V., Marambio, J. P., Wadsworth, S. & Gallardo-Escarate, C. Insights into the olfactory system of the ectoparasite Caligus rogercresseyi: Molecular characterization and gene transcription analysis of novel ionotropic receptors. Exp Parasitol 145, 99–109, https://doi.org/10.1016/j.exppara.2014.08.003 (2014).
Komisarczuk, A. Z., Grotmol, S. & Nilsen, F. Ionotropic receptors signal host recognition in the salmon louse (Lepeophtheirus salmonis, Copepoda). Plos One 12, https://doi.org/10.1371/journal.pone.0178812 (2017).
Gallardo-Escarate, C., Valenzuela-Munoz, V. & Nunez-Acuna, G. RNA-Seq Analysis Using De Novo Transcriptome Assembly as a Reference for the Salmon Louse Caligus rogercresseyi. Plos One 9, https://doi.org/10.1371/journal.pone.0092239 (2014).
Patil, V. S., Zhou, R. & Rana, T. M. Gene regulation by non-coding RNAs. Crit Rev Biochem Mol 49, 16–32, https://doi.org/10.3109/10409238.2013.844092 (2014).
Bensaoud, C., Hackenberg, M. & Kotsyfakis, M. Noncoding RNAs in Parasite-Vector-Host Interactions. Trends Parasitol 35, 715–724, https://doi.org/10.1016/j.pt.2019.06.012 (2019).
Mattick, J. S. & Rinn, J. L. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol 22, 5–7 (2015).
O’Brien, J., Hayder, H., Zayed, Y. & Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol 9, https://doi.org/10.3389/fendo.2018.00402 (2018).
LaMonte, G. et al. Translocation of Sickle Cell Erythrocyte MicroRNAs into Plasmodium falciparum Inhibits Parasite Translation and Contributes to Malaria Resistance. Cell Host Microbe 12, 187–199, https://doi.org/10.1016/j.chom.2012.06.007 (2012).
Monsanto-Hearne, V. & Johnson, K. N. miRNA Modulation of Insect Virus Replication. Curr Issues Mol Biol 34, 61–82, https://doi.org/10.21775/cimb.034.061 (2019).
Valenzuela-Miranda, D., Nunez-Acuna, G., Valenzuela-Munoz, V., Asgari, S. & Gallardo-Escarate, C. MicroRNA biogenesis pathway from the salmon louse (Caligus rogercresseyi): emerging role in delousing drug response. Gene 555, 231–241, https://doi.org/10.1016/j.gene.2014.11.008 (2015).
Gallardo-Escárate, C. et al. The Caligus rogercresseyi miRNome: Discovery and transcriptome profiling during the sea lice ontogeny. Agri Gene 4, 8–22, https://doi.org/10.1016/j.aggene.2017.03.002 (2017).
Kao, D. M. et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. Elife 5, https://doi.org/10.7554/eLife.20062 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Gregory, T. R. http://www.genomesize.com (2019).
Jeong, C.-B. et al. The genome of the harpacticoid copepod Tigriopus japonicus: Potential for its use in marine molecular ecotoxicology. Aquat Toxicol 222, https://doi.org/10.1016/j.aquatox.2020.105462 (2020).
Ghurye, J. et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. Plos Comput Biol 15, https://doi.org/10.1371/journal.pcbi.1007273 (2019).
Pal, K., Forcato, M. & Ferrari, F. Hi-C analysis: from data generation to integration. Biophys Rev 11, 67–78, https://doi.org/10.1007/s12551-018-0489-1 (2019).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295, 1306–1311, https://doi.org/10.1126/science.1067799 (2002).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–U354, https://doi.org/10.1038/Nmeth.1923 (2012).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Gene Prediction: Methods and Protocols 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Bedell, J. A., Korf, I. & Gish, W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041, https://doi.org/10.1093/bioinformatics/16.11.1040 (2000).
Bao, W. D., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA-Uk 6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res 14, 988–995, https://doi.org/10.1101/gr.1865504 (2004).
Humann, J. L., Lee, T., Ficklin, S. & Main, D. Structural and Functional Annotation of Eukaryotic Genomes with GenSAS. Gene Prediction: Methods and Protocols 1962, 29–51, https://doi.org/10.1007/978-1-4939-9173-0_3 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res 45, D190–D199, https://doi.org/10.1093/nar/gkw1107 (2017).
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32, D258–261, https://doi.org/10.1093/nar/gkh036 (2004).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–205, https://doi.org/10.1093/nar/gkt1076 (2014).
UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515, https://doi.org/10.1093/nar/gky1049 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP229458 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP212140 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP067375 (2015).
NCBI Assembly https://identifiers.org/insdc.gca:GCA_013387185.1 (2020).
Gallardo-Escárate, C. Additional annotation files_GenSAS. figshare https://doi.org/10.6084/m9.figshare.12847493 (2020).
Acknowledgements
This study was funded by the Program for Health Management of Aquaculture, FIE‐2015‐V014, FONDECYT grant #1180867, and FONDAP grant #15110027, CONICYT-Chile.
Author information
Authors and Affiliations
Contributions
C.G.-E., W.W. and S.R. designed and supervised the study. V.V., G.N.A., D.V.M. and A.T.G. prepared the samples. C.G.E., W.W., S.R., I.L., D.V.M. and B.N. analysed all sequencing data. C.G.E., W.W. and S.R. wrote the manuscript with the other authors’ helps. All authors revised the draft and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
IL and BN are employees and shareholders of Phase Genomics, Inc, a company commercializing Hi-C technology.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Gallardo-Escárate, C., Valenzuela-Muñoz, V., Nuñez-Acuña, G. et al. Chromosome-scale genome assembly of the sea louse Caligus rogercresseyi by SMRT sequencing and Hi-C analysis. Sci Data 8, 60 (2021). https://doi.org/10.1038/s41597-021-00842-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00842-w
This article is cited by
-
Sex-Biased Transcription Expression of Vitellogenins Reveals Fusion Gene and MicroRNA Regulation in the Sea Louse Caligus rogercresseyi
Marine Biotechnology (2024)
-
Silencing of the Vasa gene by RNA Interference Affects Embryonic Development and Reproductive Output in the Sea Louse Caligus rogercresseyi
Marine Biotechnology (2023)
-
Proximity ligation strategy for the genomic reconstruction of microbial communities associated with the ectoparasite Caligus rogercresseyi
Scientific Reports (2022)
