
Abstract
Sacred lotus (Nelumbo nucifera, or lotus) is one of the most widely grown aquatic plant species with important uses, such as in water gardening and in vegetable and herbal medicine. A public genomic database of lotus would facilitate studies of lotus and other aquatic plant species. Here, we constructed an integrative database: the Nelumbo Genome Database (NGD, http://nelumbo.biocloud.net). This database is a collection of the most updated lotus genome assembly and contains information on both gene expression in different tissues and coexpression networks. In the NGD, we also integrated genetic variants and key traits from our 62 newly sequenced lotus cultivars and 26 previously reported cultivars, which are valuable for lotus germplasm studies. As applications including BLAST, BLAT, Primer, Annotation Search, Variant and Trait Search are deployed, users can perform sequence analyses and gene searches via the NGD. Overall, the valuable genomic resources provided in the NGD will facilitate future studies on population genetics and molecular breeding of lotus.
Measurement(s) | reference genome data • whole genome sequencing • transcriptome |
Technology Type(s) | Hi-C • PacBio Sequel System • Illumina sequencing • RNA sequencing • DNA sequencing |
Sample Characteristic - Organism | Nelumbo nucifera |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13487271
Similar content being viewed by others

Background & Summary
Sacred lotus (Nelumbo nucifera, or lotus) is an early-diverging eudicot with important value in terms of understanding the origin and evolution of eudicots1,2. Lotus has a widespread native distribution, ranging from Asia to northern Australia, and it is one of the most economically important aquatic plant species, with widespread uses, such as in water gardening and in vegetable and herbal medicine3,4,5. In horticulture, lotus is classified into three cultivated types according to their utilization: seed lotus, flower lotus and rhizome lotus. The genome of lotus (2n = 16, assembly size = 821.2 Mb) has been sequenced and assembled, providing an unprecedented opportunity for genetic studies and molecular breeding of lotus6,7,8.
Since the first draft genome assembly of the lotus variety China Antique was released9, many genomic studies have been carried out on lotus, such as whole-genome resequencing7,10,11, transcriptomic12,13,14, miRNA-based15,16,17 and gene family studies18,19. The recent chromosome-level assembly of the China Antique genome facilitates genome-wide studies of functional genes and the evolution of lotus, but a web-based public database of lotus is still unavailable8. Due to public demand for an integrative genomic resource of lotus, we report our Nelumbo Genome Database (NGD, nelumbo.biocloud.net), which comprehensively houses, processes and integrates the newest assembly of lotus variety China Antique, the expression profiles of various tissues, genetic variants and phenotypes of 88 key lotus cultivars (Fig. 1).

A schematic of the data collection and utilities for the Nelumbo Genome Database (NGD).
Major datasets
The NGD is a collection of the new lotus assembly and annotations8, of which 34,481 genes harbor complete ORFs (> = 30 aa). A total of 28,676 genes were defined as high-confidence genes, with 14,991, 20,878, 15,276, 5,924, 29,095, 20,325, and 28,310 annotated genes in the KOG, PFAM, GO, KEGG, Nr, SwissProt and TrEMBL databases, respectively (Table 1). The NGD also houses the sequences of 150,589 unique transcript isoforms based on RNA-seq and PacBio SMRT methods from our previous studies14,17. It also contains the sequences of 1,517 lotus transcription factors (TFs), which are classified into 56 TF (sub)families. Furthermore, sequence data of lotus gene families, transposable elements (TEs) and other repeats are also present in the NGD, and information concerning gene expression levels and highly coexpressed genes from data from a coexpression (WGCNA) network based on 69 RNA-seq samples from 11 lotus tissues (seed coat, cotyledon, receptacle, carpel, stamen, petal, rhizome, leaf, root, petiole and apical bud tissues) is present in the NGD. Furthermore, information concerning a total of 26,939,834 high-quality SNPs (single nucleotide polymorphisms), 4,177,974 InDels and key horticultural traits from 88 lotus cultivars is present in the NGD.
Uses
Through data collection and downstream processing, our platform provides the most complete lotus genome assembly for browsing via GBrowse or JBrowse20,21. Genes, DNA sequences, amino acids, SNPs and InDels can be viewed via GBrowse. The gene information page includes gene-splicing structures, sequences, and functional annotations such as those from the PFAM, KEGG, GO, KOG, SwissProt, TrEMBL and Nr databases. RNA-seq-based expression profiles across different tissues are also retrievable and can be visualized via a heatmap. Searching genes by keywords is also possible in the NGD. Additionally, coexpressed genes of a query gene in the WGCNA-derived network can be retrieved by setting a weight threshold; these coexpressed genes are likely involved in the same biological process as the query gene. Coding sequences and genomic sequences can be searched based on sequence similarity via BLAST or BLAT. Primers for the PCR experiments can also be designed directly in the NGD.
Methods
Data processing
Gene predictions on our chromosome-level genome assembly were performed using transcriptomes, gene homology and ab initio identification. A list of publicly available RNA-seq datasets, which mainly contain samples of the China Antique variety, were downloaded from the NCBI SRA database (Online-only Table 1). First, the corrected consensus PacBio full-length transcripts were mapped to the lotus reference genome using GMAP14. RNA-seq reads (Illumina) were then mapped to the genome using the HISAT2-StringTie pipeline22. All the transcripts were further merged using TACO23. Coding DNA sequences (CDSs) of transcripts were predicted using Transdecoder (https://github.com/TransDecoder). Second, homology-based gene prediction was performed using GeMoMa, which used genome sequences and gene coordinates from Arabidopsis thaliana24, Carica papaya25, Vitis vinifera26, Macadamia ternifolia (Proteales)27 and Brachypodium distachyon28 as inputs. Third, ab initio prediction was performed using Braker2, which used transcript coordinates of RNA-seq as a guide29. Finally, all predictions were merged, and for each gene with more than one gene model (transcripts), the longest one was chosen as the representative gene model. Genes with an ORF less than 30 aa were discarded. Further, high-confidence gene sets were defined as those whose genes that either were homologous to those in other plant species in Plant Plaza 4.0 (https://bioinformatics.psb.ugent.be/plaza/versions/plaza_v4_dicots/) or were supported by RNA-seq (FPKM > 0.1). To quantify the expression of each gene in different lotus tissue samples, FPKM values across different RNA-seq samples were obtained via StringTie22. A coexpression network of different genes based on the expression profile was constructed using the WGCNA (v1.0) package in R30. Specifically, genes with an average FPKM > 0.1 and a coefficient of variation (CV) of gene expression (FPKM) > 2 were retained for the WGCNA. Genes were clustered hierarchically based on Topological Overlap Matrix31 and were assigned to nine modules (minimum module size of 600 and minimum module similarity of 0.5). The weight values between genes were used to represent the connectivity between genes.
Gene functions were annotated using the Gene Ontology (GO), KEGG, KOG, Pfam, SwissProt, TrEMBL, and Nr databases via KOBAS 3.0, BlastKOALA, PfamScan and BLAST32,33. As protein domains are conserved units shared by related genes, we clustered genes into domain families (gene families) according to the HMM Pfam domain annotations34. In addition, all transcription factors (TFs) were predicted and clustered into TF families via PlantTFDB 4.035.
There were 88 recorded lotus cultivars chosen in this study as representing various floral traits (color, shape, flowering time, etc.). Among these cultivars, 62 were sequenced in our current study, while the sequences of 26 with detailed phenotypic records were downloaded from the NCBI database (Online-only Table 2). Genomic DNA was first extracted from young leaves by the CTAB method36, and then DNA libraries were constructed by cutting the DNA into 250~280 bp fragments using a NEBNext® Ultra DNA Library Prep Kit for Illumina (NEB, USA) following the manufacturer’s recommendations. Paired-end reads (PE150) were sequenced on an Illumina NovaSeq. 6000 (San Diego, CA, USA), which generated approximately 16 × -depth data for each cultivar sample. Clean reads were obtained by removing the adapters and low-quality reads, including those comprising > 10% N, with < 20% low-quality bases, with low-quality/ambiguous fragments at the read ends within a 5 bp window and with a quality < 20 via FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). The clean reads were mapped to the reference genome by BWA-men37. Variants were subsequently called by pipeline via GATK 4.0 (Genome Analysis Toolkit) with further SNP hard-filtering parameters (“QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0” and InDel hard-filtering parameters of “QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0”)38. The phenotypes and some images of these cultivars were collected from reference books39,40; the phenotypes were further validated across two years of field investigations. Images of floral traits for these cultivars displayed in the NGD were taken mostly during flowering at the Wuhan Institute of Landscape Architecture (Wuhan, China).
Database construction
All genomic sequence, annotation, expression, and genetic variation data were stored via MySQL on a Ubuntu server. A user-friendly website was developed using HTML5 and JavaScript; this website which can be accessed through different browsers, such as Google Chrome and Firefox. Gene models and transcript isoforms are provided via GBrowse and JBrowse. Heatmaps of gene expression are plotted via R, and query searches are achieved via JavaScript and Java. Common utilities for genomic studies such as BLAST, BLAT and Primer Design are also deployed and accessible.
Data Records
The genomic raw PacBio sequencing data are available in the NCBI Sequence Read Archive (SRA) database under accession numbers SRR754912941 and SRR754913042, and the Illumina and Hi-C sequencing data are deposited under SRR761555343 and SRR763152344, which helped us in our genome assembly (Online-only Table 1). Raw whole-genome resequencing reads for 62 strains can be downloaded from the NCBI database under Bioproject accession SRP17354745, and the resequencing data of the other 22 cultivars are also accessible via the NCBI SRA46,47 (Online-only Table 2). The latest assembly and annotations of the ‘China Antique’ lotus variety is deposited in the Nelumbo Genome Database (Download links: http://nelumbo.biocloud.net/downloadData/download?path = NNU.genomic.fa and http://nelumbo.biocloud.net/downloadData/download?path = NNU.gff3). Additionally, this Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession DUZY00000000, and the version described in this paper is version DUZY0100000048. Improved gene and repeat (including transposable element) predictions (GFF3), coding and peptide sequences (FASTA), gene and transcript functional annotations, gene expression and coexpression profiles, SNP and InDel variations, phenotypic traits and images for 88 lotus strains have been uploaded into the Figshare database49 and deployed in the newly developed Nelumbo Genome Database (http://nelumbo.biocloud.net).
Technical Validation
Quality control of genome annotation, expression, and genome resequencing was performed during data processing for the NGD.
Genome annotation
We used a set of conserved single-copy plant genes from the BUSCO database to assess the completeness of gene annotations50. Compared with previous annotations of the lotus variety China Antique (BUSCOs = 74.6%), our new annotation version provides much improved, complete BUSCOs (97.5%), and 41,140 out of 46,713 annotated genes with either complete or partial ORFs (88%) were validated by 69 transcriptome datasets (Figure S1a), which suggests relatively high quality and completeness of the genome assembly and gene annotations (Table 2).
Gene expression
To ensure that the FPKMs of genes accurately reflect the gene expression in different tissues and at different developmental stages, hierarchical clustering of gene FPKMs across different RNA-seq samples of the variety China Antique was performed via Expander 6.051. All sample repeats clustered together, while all developmental stages from the same tissue clustered together, except for one petiole sample, and the relative expression of randomly selected genes in different tissues was validated through qRT-PCR (Fig. 2 and Figure S1b). Therefore, we confirmed the accuracy of FPKM as an indicator of lotus gene expression.

Hierarchical clustering of different RNA-seq samples based on a gene expression matrix (FPKM) from the lotus variety China Antique. Note that only a small portion of the genes are shown in the heatmap.
Whole-genome resequencing
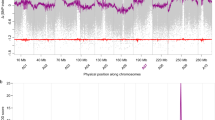
Before genome mapping, adapters and low-quality Illumina reads were filtered and removed (see the Methods). Base content, error rate, insert size distribution and log-transformed read coverage across the lotus chromosomes were checked, all of which met the criteria for downstream analyses (Fig. 3). The quality of genome mapping was also checked. The average mapping rate for cultivars from this study was 99.18%, while the numbers in the other cultivars collected from two previous studies were 98.98% and 99.13% (Online-only Table 2). The average depth for the cultivars from the current study was 16.1×, while the depth was 12.4× and 11.8× for cultivars from the other two reports (Online-only Table 2). To ensure the final quality of SNPs and InDels called by the GATK pipeline, stringent hard-filtering parameters were set (see the Methods). Because the majority of alleles in the SNP data set are expected to be shared by at least two individuals, we plotted the frequency of SNPs according to the minor allele count (MAC) across the 88 cultivars. Indeed, most of the SNPs had MACs ≥ 2, while the SNP density peaked around MACs of four or five (Fig. 4). SNP variants were further validated and visualized using IGVtools (http://software.broadinstitute.org/software/igv/igvtools) (Figure S1c).

Quality evaluation of the genome resequencing data of cultivars, including the base content (a), error rate (b), insert size distribution (c) and log-transformed read coverage across eight lotus chromosomes (d), as demonstrated by the example of lotus cultivar Xiaoxia. The resequencing quality met the criterion for downstream variant calling analyses.

Distribution of SNPs according to the minor allele count (MAC) across 88 lotus cultivars.
Code availability
The genomic and transcriptomic sequence data were produced by corresponding software provided by the sequencing platform manufacturer, and the software (including versions, parameters and settings) used for genome assembly was cited in the Methods section, with default parameters used when no detailed parameters were mentioned. The code for the NGD construction in Java is available in the Figshare database49.
References
Gandolfo, M. A., Nixon, K. C. & Crepet, W. L. Cretaceous flowers of Nymphaeaceae and implications for complex insect entrapment pollination mechanisms in early angiosperms. Proc. Natl. Acad. Sci. USA 101, 8056–8060 (2004).
Zheng, C. & Sankoff, D. Practical halving; the Nelumbo nucifera evidence on early eudicot evolution. Comput Biol Chem 50, 75–81 (2014).
Hayes, V., Schneider, E. L. & Carlquist, S. Floral development of Nelumbo nucifera (Nelumbonaceae). Int. J. Plant Sci. 161, S183–S191 (2000).
Slocum, P. D. Waterlilies and Lotuses: Species, Cultivars, and New Hybrids. (Timber Press, 2005).
Zhou, M. et al. Identification and comparison of anti-inflammatory ingredients from different organs of lotus nelumbo by UPLC/Q-TOF and PCA coupled with a NF-kappaB reporter gene assay. PloS One 8, e81971 (2013).
Cheng, T. et al. Development and identification of three functional markers associated with starch content in lotus (Nelumbo nucifera). Sci. Rep. 10, 4242 (2020).
Li, Y. et al. Comparative population genomics reveals genetic divergence and selection in lotus. Nelumbo nucifera. BMC Genom 21, 146 (2020).
Shi, T. et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol. Biol. Evol. 37, 2394–2413 (2020).
Ming, R. et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol 14, R41 (2013).
Huang, L. et al. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera). J Integr Plant Biol 60, 2–15 (2018).
Zhao, M. et al. Detection of highly differentiated genomic regions between lotus (Nelumbo nucifera Gaertn.) with contrasting plant architecture and their functional relevance to plant architecture. Front. Plant Sci. 9, 1219 (2018).
Yang, M. et al. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical Lotus (Nelumbo nucifera). Sci. Rep. 5, 13059 (2015).
Li, J. et al. Systematic transcriptomic analysis provides insights into lotus (Nelumbo nucifera) seed development. Plant Growth Regul 86, 339–350 (2018).
Zhang, Y., Nyong, A. T., Shi, T. & Yang, P. The complexity of alternative splicing and landscape of tissue-specific expression in lotus (Nelumbo nucifera) unveiled by Illumina- and single-molecule real-time-based RNA-sequencing. DNA Res 26, 301–311 (2019).
Zheng, Y. et al. Genome-wide analysis of microRNAs in sacred lotus, Nelumbo nucifera (Gaertn). Tropical Plant Biol. 6, 117–130 (2013).
Shi, T., Wang, K. & Yang, P. The evolution of plant microRNAs: insights from a basal eudicot sacred lotus. Plant J 89, 442–457 (2017).
Zhang, Y., Rahmani, R. S., Yang, X., Chen, J. & Shi, T. Integrative expression network analysis of microRNA and gene isoforms in sacred lotus. BMC Genom 21, 429 (2020).
Wang, Y. et al. Genome-wide identification and characterization of GRAS transcription factors in sacred lotus (Nelumbo nucifera). PeerJ. 4, e2388 (2016).
Li, H., Yang, X., Lu, M., Chen, J. & Shi, T. Gene expression and evolution of Family-1 UDP-glycosyltransferases—insights from an aquatic flowering plant (sacred lotus). Aquat. Bot. 166, 103270 (2020).
Chui, R., Jaromczyk, J. W., Moore, N. & Schardl, C. L. FPD2GB2: automating a transition from a customized genome browser to GBrowse2. BMC Bioinform 14, A17 (2013).
Buels, R. et al. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol 17, 66 (2016).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Niknafs, Y. S., Pandian, B., Iyer, H. K., Chinnaiyan, A. M. & Iyer, M. K. TACO produces robust multisample transcriptome assemblies from RNA-seq. Nat. Methods. 14, 68–70 (2017).
Michael, T. P. et al. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9, 541 (2018).
Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature. 452, 991–996 (2008).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 449, 463–467 (2007).
Nock, C. J., Baten, A. & King, G. J. Complete chloroplast genome of Macadamia integrifolia confirms the position of the Gondwanan early-diverging eudicot family Proteaceae. BMC Genom 15, S13 (2014).
Fox, S. E. et al. Sequencing and de novo transcriptome assembly of Brachypodium sylvaticum (Poaceae). Appl. Surf. Sci. 1, apps.1200011 (2013).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. Whole-genome annotation with BRAKER. Methods Mol. Biol. 1962, 65–95 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9, 559 (2008).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005).
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y. & Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res 32, D277–280 (2004).
Ai, C. & Kong, L. CGPS: A machine learning-based approach integrating multiple gene set analysis tools for better prioritization of biologically relevant pathways. J Genet Genomics 45, 489–504 (2018).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res 47, D427–D432 (2019).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45, D1040–D1045 (2017).
Cota-Sánchez, J. H., Remarchuk, K., Ubayasena, K. & Ready-to-use, D. N. A. extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 24, 161–167 (2006).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26, 589–595 (2010).
McKenna, A. et al. The genome analysis toolkit: a mapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Zhang, X. & Wang, Q. New Lotus Flower Cultivars in China. (China Forestry Publishing House, 2011).
Zhang, X. & Wang, Q. Lotus Flower Cultivars in China. (China Forestry Publishing House, 2005).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRR7549129 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRR7549130 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRR7615553 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRR7631523 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP173547 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP145546 (2018).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP090666 (2016).
GenBank whole genome shotgun sequencing project https://identifiers.org/ncbi/insdc:DUZY00000000 (2020).
Li, H. Nelumbo genome database, an integrative resource for gene expression and variants of Nelumbo nucifera. figshare https://doi.org/10.6084/m9.figshare.c.5108198 (2020).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Sharan, R., Maron-Katz, A. & Shamir, R. CLICK and EXPANDER: a system for clustering and visualizing gene expression data. Bioinformatics. 19, 1787–1799 (2003).
Acknowledgements
This work was supported by grants from the Strategic Priority Research Program CAS (No. XDB31000000), the National Natural Science Foundation of China (No. 31570220, No. 31870208 and No. 31700197), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (No. 2019335), the Bureau of Landscaping and Forestry of Wuhan Municipality (No. WHGF2019A10), the Hubei Provincial Natural Science Foundation of China (No. 2019CFB275), and the Hubei Chenguang Talented Youth Development Foundation. We thank Razgar Seyed Rahmani from Ghent University for the discussion.
Author information
Authors and Affiliations
Contributions
Genome sequencing, assembly and annotation: T.S.; RNA-seq and gene expression: Y.Z., X.Y.; genome resequencing of cultivars: H.L., X.Y.; phenotype collection: X.Y., Z.G.; sample collection and experiments: C.W., Y.L.; communications, web design and conceptualization: J.C.; manuscript writing and revising: H.L., J.C. and T.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Online-only Tables
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Li, H., Yang, X., Zhang, Y. et al. Nelumbo genome database, an integrative resource for gene expression and variants of Nelumbo nucifera. Sci Data 8, 38 (2021). https://doi.org/10.1038/s41597-021-00828-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00828-8
This article is cited by
-
Lotus (Nelumbo nucifera): a multidisciplinary review of its cultural, ecological, and nutraceutical significance
Bioresources and Bioprocessing (2024)
-
The slow-evolving Acorus tatarinowii genome sheds light on ancestral monocot evolution
Nature Plants (2022)
