
Abstract
Shift work is known to be associated with an increased risk of neurological and psychiatric diseases, but how it contributes to the development of these diseases remains unclear. Chronic jet lag (CJL) induced by shifting light-dark cycles repeatedly is a commonly used protocol to mimic the environmental light/dark changes encountered by shift workers. Here we subjected wildtype mice to CJL and performed positron emission tomography imaging of glucose metabolism to monitor brain activities. We also conducted RNA sequencing using prefrontal cortex and nucleus accumbens tissues from these animals, which are brain regions strongly implicated in the pathology of various neurological and psychiatric conditions. Our results reveal the alterations of brain activities and systematic reprogramming of gene expression in brain tissues under CJL, building hypothesis for how CJL increases the susceptibility to neurological and psychiatric diseases.
Measurement(s) | brain measurement • sleep duration • sleep quality • fragments per kilobase of exon per million mapped reads |
Technology Type(s) | positron emission tomography • animal activity monitoring system • RNA sequencing |
Factor Type(s) | chronic jet lag |
Sample Characteristic - Organism | Mus musculus |
Sample Characteristic - Environment | laboratory environment |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12924248
Similar content being viewed by others


Background & Summary
Approximately 15%–30% of the working population worldwide are engaged in some type of shift work1. Based on epidemiological surveys, shift work is associated with numerous adverse health outcomes such as type 2 diabetes, cancer, cardiovascular disorders and immune dysfunction2,3. Shift work is also known to increase the risk of neurological diseases. To be specific, shift work may trigger migraine, exacerbate epilepsy and increase dementia incidence4,5,6. In addition, shift work is associated with cognitive deficits7,8,9, sleep disorders1,10,11, mood alteration and increases the risk of mental illnesses including depression, anxiety, alcohol abuse, and schizophrenia12,13,14,15. Individuals undergoing shift work experience alternating light/dark cycles for extended time and suffer from circadian disruptions due to the misalignment between endogenous rhythm and external time. To mimic shift work condition, a number of studies used rodents subjected to chronic jet lag (CJL) by continuously shifting the timing of light-dark cycles and found that CJL promotes tumour progression16,17,18, obesity19, addiction20, and impairs innate immune responses21. Recent work in rodents also demonstrated that CJL leads to phenotypes related to mood disorders, but the mechanism is largely unknown22,23.
Glucose turnover rate serves as an indicator of brain activities, while alterations of brain activities are linked to many neurological and psychiatric diseases24. Abnormal glucose metabolism in the brain has been reported in dementia, epilepsy, major depressive disorder, and bipolar disorder25,26,27. The prefrontal cortex (PFC) is known to be involved in regulating a number of cognitive and emotional processes28. Dysfunction of the PFC has been found in various psychiatric and neurological disorders, including depression, anxiety disorders, addiction, schizophrenia, autism spectrum disorders, Alzheimer’s disease, and Parkinson’s disease29,30,31,32. Nucleus accumbens (NAc) plays a central role in processing motivation, reward and aversion33,34,35. NAc has been implicated in the pathophysiology and treatment of mental illnesses such as major depressive disorder, addictive disorders, schizophrenia, obsessive-compulsive disorder and anorexia nervosa36,37,38,39.
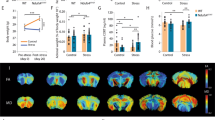
In this study, we subjected wildtype mice to CJL following a previously reported protocol40 (Fig. 1). Total sleep duration is not reduced by CJL, indicating that the physiological and molecular changes we report here are not due to deficient sleep caused by CJL (Fig. 2). We employed positron emission tomography (PET) to monitor glucose uptake in the brain of mice maintained under baseline condition or CJL for slightly over a month, as it has previously been shown that CJL treatment for about a month can elicit prominent changes in metabolic indices and lung mechanics40,41. We present raw PET images as well as quantified data collected on Day 34 of CJL treatment, which demonstrate that CJL significantly reduces activities throughout the brain (Fig. 3). Given the prominent and pervasive alterations in brain activity observed by Day 34 of CJL, we reasoned that molecular changes contributing to this should start to occur at an earlier stage of CJL treatment. It has been reported that metabolic phenotypes start to become significant by Day 10 of CJL, and ten days of CJL is sufficient to induce substantial effects on the expression of a number of circadian genes40,42. Therefore, we conducted RNA sequencing (RNA-seq) on PFC and NAc harvested from animals during the light and dark phase under baseline condition and on Day 10 of CJL treatment, respectively. These data will provide valuable information for studies that investigate how shift work or circadian disruptions increase the risk of disorders associated with the brain.

Light regime for chronic jet lag treatment. Male wildtype mice were exposed to stable or shiftwork-like light conditions. Black bars represent dark phase and hollow bars represent light phase. Solid triangles and square brackets indicate sampling time of RNA-seq and time of PET imaging, respectively. Ctrl, control; CJL, chronic jet lag.

Chronic jet lag does not reduce sleep duration. Average percent sleep (a), the number of sleep bout in 24 hours (b) and average bout length in seconds (c) of male wildtype mice on the 8th, 18th, and 28th day of CJL (N = 7). Data are presented as mean ± SEM, Mann-Whitney test, *P < 0.05, **P < 0.01. Ctrl, control; CJL, chronic jet lag.

Chronic jet lag reduces brain glucose metabolism. Bars indicate standard uptake value (SUV) of glucose in the entire brain and different brain regions of control mice (N = 6) and mice on the 34th day of CJL treatment (N = 9). Data are presented as mean ± SEM, Mann-Whitney test, *P < 0.05, **P < 0.01, ***P < 0.001. Ctrl, control; CJL, chronic jet lag; RSTR, right striatum; LSTR, left striatum; CTX, cerebral cortex; RHIP, right hippocampus; LHIP, left hippocampus; THA, thalamus; CB, cerebellum; BFS, basal forebrain septum; HYP, hypothalamus; RAMY, right amygdale; LAMY, left amygdale; BS, brain stem; CG, central gray; SC, superior colliculus; OLF, olfactory bulb; RMID, right midbrain; LMID, left midbrain; LIC, left inferior colliculus; RIC, right inferior colliculus.
Methods
Animals and chronic jet lag treatment
8-week-old male C57BL/6 J mice purchased from Model Animal Research Center of Nanjing University were assigned to different cages randomly with approximately 5 animals per cage. Animals were housed in light-tight cabinets with time-controlled illumination. Food and sterilized water were accessible ad libitum. The mice were housed under 12 hour light: 12 hour dark (12L12D) for 2 weeks for entrainment prior to CJL treatment. The experimental design is displayed in Fig. 1. For CJL group, lights on and off times were advanced by 6 hours every 2 days following a previously published protocol, whereas for control group they remain unchanged40,43. Nine mice were subjected to PET imaging on Day 34 of CJL (the last jetlag event before PET imaging occurred on Day 32) along with six controls. The brain tissues of six mice (three per time point) were harvested for RNA-seq on Day 10 of CJL (the last jetlag event before tissue collection occurred on Day 8) along with equal number of controls. To assess sleep, seven mice were individually housed and sleep monitoring was conducted on Day 8, 18, and 28 of CJL. All mouse work was performed in accordance with the guidelines of Institutional Animal Care and Use Committee at Huazhong University of Science and Technology.
Sleep monitoring
Sleep was monitored by PiezoSleep Mouse Behaviour Tracking System as described earlier44. Briefly, seven animals were individually housed in a cage with piezoelectric film sensor underneath that transmitted activity signals (motions and breathing movements) to monitoring software (PiezoSleep 2.18, Signal Solutions). Sleep can be distinguished from wakefulness by rhythmic signals (approximately 3 Hz) generated by typical respiration patterns during sleep44. The baseline pressure signal from the piezoelectric sensors was sampled at 120 Hz. Features associated with sleep and wake behaviors were extracted from the pressure signals and saved every 2 seconds. Sleep-wake classification was performed automatically based on decision statistics45. Data collected were binned over 1 hour using the average of percent sleep (average percent of time in sleep state). Data were analyzed with SleepStats and exported as hourly percent sleep, hourly sleep bout number and hourly average sleep bout length of the specified day. Sleep bout was computed as the duration of an interval of continuous sleep. The minimum sleep bout length parameter was set to 30 s by default, which means a bout length count was initiated only when a 30 s-interval contained greater than 50% sleep46,47. Setting the minimum bout length as 30 s can eliminate the impact of short and ambiguous arousals on computing bout length and reduce the probability of error46. Based on the exported data, average percent sleep, sleep bout number, and average length of sleep bout of a specified day were calculated.
PET
18F-fluorodeoxyglucose (18F-FDG) is the most commonly used marker for glucose metabolism in PET imaging48. Nine male C57BL6/J mice subjected to CJL and six control mice were used. Mice were fasted during the dark phase for 12 h before the test. Prior to PET imaging, animals were injected with (250 ± 10 μCi) 18F-FDG intraperitoneally. After one hour, they were anesthetized with 2% isoflurane and imaging was conducted from Zeitgeber Time 1 (ZT1, 1 hour after lights on; ZT0 is defined as the time of lights on) to ZT6. Images of mouse brains were obtained with the static scanning pattern (10 min) by Trans-PET®BioCaliburn® LH (Raycan Technology, China), a PET system for imaging small animals49. The PET images were reconstructed using three-dimensional OSEM method with a voxel size of 0.5 × 0.5 × 0.5 mm3. Glucose uptake was measured as the mean standardized uptake value (SUV).
Brain dissection and RNA extraction
Brain tissues of three CJL treated animals and three control animals fed ad libitum were harvested at ZT1 and ZT13, respectively. After cervical dislocation, the skin was flipped over the eyes to free the skull. Then the skull was broken and brain was removed gently out of the skull. The brain was immediately transferred to a Petri dish chilled on ice and placed with ventral surface facing up. The brains of mice were dissected according to previously published protocols50,51,52,53. Coronal sections were made from the rostral end of the brain using a sharp and chilled razor blade. PFC, the anterior part of the brain just behind the olfactory bulb, was removed from the first ~1.5 mm-thick coronal section50,51. NAc was located in the subsequent section (~1.0 mm-thick) and was identified based on the location of anterior commissure with the approximate anterior-posterior coordinate of +1.8 mm~0.6 mm from Bregma52,53. The dissected PFC and NAc tissues were immediately transferred to liquid nitrogen and stored at −80 °C for further processing.
Total RNA was extracted using RNA isolater (Vazyme, China). RNA integrity was assessed with RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Alilent Technologies, USA) before the samples were sent for RNA-seq.
Library preparation, RNA sequencing and data processing
The sequencing libraries were generated using VAHTS mRNA-seq v2 Library Prep Kit for Illumina® (Vazyme, NR601) following manufacturer’s recommendations. Briefly, mRNA was purified and fragmented and cDNA was synthesized with the mRNA fragments as templates. Then the cDNAs were ligated with special sequencing adaptor and the products with appropriate size were selected for PCR. After passing quality examination, the generated libraries were sequenced on Illumina Hiseq X Ten platform with 150 bp paired-end module.
The raw reads were filtered to produce clean data by excluding sequencing adaptors and low quality reads, including reads containing over 5% “N” and those containing >50% bases with quality value less than 10. The quality assessment of clean data was performed using FastQC v0.11.9.
Sequencing reads were then aligned to the mouse mm10 reference genome using HISAT2. The differential expression analysis was performed by DESeq. 2 with default parameters. The significance threshold of P < 0.05 was applied. The number of differentially expressed genes (DEGs) and the number of genes that exhibited a fold-change >2 in each brain tissue at each time point were displayed in Table 1.
Technical Validation
A high percentage of clean reads from each sample was acquired after filtering the raw reads. The statistics summary of clean reads is presented in Table 2. All samples produced more than 98.5% clean reads and >93.60% of the clean data were mapped to the reference genome. In addition, GC content, Q20 and Q30 were also calculated. For all samples, the GC content was stable with distributed range from 48.36% to 49.18%. The read base quality was assessed by FastQC and displayed in Fig. 4. Most of the base quality scores were above 30, suggesting high-quality RNA-seq data.

FastQC report for the assessment of RNA sequencing quality. (a,b) The quality scores per base along the reads from one sample. The yellow box represents the interquartile range (25–75%). The upper and lower whiskers represent the 90% and 10% points, respectively. The blue line indicates mean value. (c,d) The distribution of mean sequence quality scores for all reads from one sample.
References
Boivin, D. B. & Boudreau, P. Impacts of shift work on sleep and circadian rhythms. Pathol. Biol. 62, 292–301 (2014).
Kecklund, G. & Axelsson, J. Health consequences of shift work and insufficient sleep. BMJ 355, i5210 (2016).
Evans, J. A. & Davidson, A. J. Health consequences of circadian fisruption in humans and animal models. Prog. Mol. Biol. Transl. Sci. 119, 283–323 (2013).
Kuo, W. Y. et al. Higher migraine risk in healthcare professionals than in general population: a nationwide population-based cohort study in Taiwan. J. Headache Pain 16, 102 (2015).
Ladou, J. Health effects of shift work. West. J. Med. 137, 525–530 (1982).
Bokenberger, K. et al. Shift work and risk of incident dementia: a study of two population-based cohorts. Eur. J. Epidemiol. 33, 977–987 (2018).
Cho, K., Ennaceur, A., Cole, J. C. & Suh, C. K. Chronic jet lag produces cognitive deficits. J. Neurosci. 20, RC66 (2000).
Cho, K. W. Chronic ‘jet lag’ produces temporal lobe atrophy and spatial cognitive deficits. Nat. Neurosci. 4, 567–568 (2001).
Marquie, J. C., Tucker, P., Folkard, S., Gentil, C. & Ansiau, D. Chronic effects of shift work on cognition: findings from the VISAT longitudinal study. Occup. Environ. Med. 72, 258–264 (2015).
Ohayon, M. M., Lemoine, P., Arnaud-Briant, V. & Dreyfus, M. Prevalence and consequences of sleep disorders in a shift worker population. J. Psychosom. Res. 53, 577–583 (2002).
Akerstedt, T. Shift work and disturbed sleep/wakefulness. Occup. Med. 53, 89–94 (2003).
Kalmbach, D. A., Pillai, V., Cheng, P., Arnedt, J. T. & Drake, C. L. Shift work disorder, depression, and anxiety in the transition to rotating shifts: the role of sleep reactivity. Sleep Med. 16, 1532–1538 (2015).
Lee, A. et al. Night shift work and risk of depression: meta-analysis of observational studies. J. Korean Med. Sci. 32, 1091–1096 (2017).
Scott, A. J., Monk, T. H. & Brink, L. L. Shiftwork as a risk factor for depression: a pilot study. Int. J. Occup. Environ. Health 3, S2–S9 (1997).
Brown, J. P. et al. Mental health consequences of shift work: an updated review. Curr. Psychiatry Rep. 22, 7 (2020).
Filipski, E. et al. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 64, 7879–7885 (2004).
Filipski, E., Li, X. & Levi, F. Disruption of circadian coordination and malignant growth. Cancer Causes Control 17, 509–514 (2006).
Filipski, E. & Levi, F. Circadian disruption in experimental cancer processes. Integr. Cancer Ther. 8, 298–302 (2009).
Oike, H., Sakurai, M., Ippoushi, K. & Kobori, M. Time-fixed feeding prevents obesity induced by chronic advances of light/dark cycles in mouse models of jet-lag/shift work. Biochem. Biophys. Res. Commun. 465, 556–561 (2015).
Doyle, S. E., Feng, H. T., Garber, G., Menaker, M. & Lynch, W. J. Effects of circadian disruption on methamphetamine consumption in methamphetamine-exposed rats. Psychopharmacology 232, 2169–2179 (2015).
Castanon-Cervantes, O. et al. Dysregulation of inflammatory responses by chronic circadian disruption. J. Immunol. 185, 5796–5805 (2010).
Horsey, E. A. et al. Chronic jet lag simulation decreases hippocampal neurogenesis and enhances depressive behaviors and cognitive deficits in adult male rats. Front. Behav. Neurosci. 13, 272 (2020).
Otsuka, T. et al. Adverse effects of circadian disorganization on mood and molecular rhythms in the prefrontal cortex of mice. Neuroscience 432, 44–54 (2020).
Segobin, S. et al. FDG-PET contributions to the pathophysiology of memory impairment. Neuropsychol. Rev. 25, 326–355 (2015).
Scholl, M., Damian, A. & Engler, H. Fluorodeoxyglucose PET in neurology and psychiatry. PET cli. 9, 371–390 (2014).
Hurwitz, T. A. et al. Regional cerebral glucose metabolism in major depressive disorder. Can. J. Psychiatry 35, 684–688 (1990).
Hosokawa, T., Momose, T. & Kasai, K. Brain glucose metabolism difference between bipolar and unipolar mood disorders in depressed and euthymic states. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 33, 243–250 (2009).
Etkin, A., Egner, T. & Kalisch, R. Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn. Sci. 15, 85–93 (2011).
Xu, P., Chen, A., Li, Y. P., Xing, X. Z. & Lu, H. Medial prefrontal cortex in neurological diseases. Physiol. Genomics 51, 432–442 (2019).
Gunaydin, L. A. & Kreitzer, A. C. Cortico-basal ganglia circuit function in psychiatric disease. Annu. Rev. Physiol. 78, 327–350 (2016).
Belleau, E. L., Treadway, M. T. & Pizzagalli, D. A. The impact of stress and major depressive disorder on hippocampal and medial prefrontal cortex morphology. Biol. Psychiatry 85, 443–453 (2019).
Murrough, J. W. et al. Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum. Brain Mapp. 37, 3214–3223 (2016).
Floresco, S. B. The nucleus accumbens: an interface between cognition, emotion, and action. Annu. Rev. Psychol. 66, 25–52 (2015).
Goto, Y. & Grace, A. A. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 31, 552–558 (2008).
Volkow, N. D. & Morales, M. The brain on drugs: from reward to addiction. Cell 162, 712–725 (2015).
Nauczyciel, C. et al. The nucleus accumbens: a target for deep brain stimulation in resistant major depressive disorder. J. Mol. Psychiatry 1, 17 (2013).
Seifert, C. L. et al. Reduced volume of the nucleus accumbens in heroin addiction. Eur. Arch. Psychiatry Clin. Neurosci. 265, 637–645 (2015).
Ballmaier, M. et al. Thought disorder and nucleus accumbens in childhood: a structural MRI study. Psychiatry Res. 130, 43–55 (2004).
Figee, M. et al. Dysfunctional reward circuitry in obsessive-compulsive disorder. Biol. Psychiatry 69, 867–874 (2011).
Casiraghi, L. P., Alzamendi, A., Giovambattista, A., Chiesa, J. J. & Golombek, D. A. Effects of chronic forced circadian desynchronization on body weight and metabolism in male mice. Physiol. Rep. 4, e12743 (2016).
Hadden, H., Soldin, S. J. & Massaro, D. Circadian disruption alters mouse lung clock gene expression and lung mechanics. J. Appl. Physiol. 113, 385–392 (2012).
Iwamoto, A., Kawai, M., Furuse, M. & Yasuo, S. Effects of chronic jet lag on the central and peripheral circadian clocks in CBA/N mice. Chronobiol. Int. 31, 189–198 (2014).
Casiraghi, L. P., Oda, G. A., Chiesa, J. J., Friesen, W. O. & Golombek, D. A. Forced desynchronization of activity rhythms in a model of chronic jet lag in mice. J. Biol. Rhythms 27, 59–69 (2012).
Mang, G. M. et al. Evaluation of a piezoelectric system as an alternative to electroencephalogram/electromyogram recordings in mouse sleep studies. Sleep 37, 1383–1392 (2014).
Sethi, M. et al. Increased fragmentation of sleep-wake cycles In the 5xFAD mouse model of Alzheimer’s disease. Neuroscience 290, 80–89 (2015).
Duncan, M. J. et al. Effects of the dual orexin receptor antagonist DORA-22 on sleep in 5XFAD mice. Alzheimer’s & Dementia 5, 70–80 (2019).
Paulose, J. K., Wang, C., O’Hara, B. F. & Cassone, V. M. The effects of aging on sleep parameters in a healthy, melatonin-competent mouse model. Nat. Sci. Sleep 11, 113–121 (2019).
Newberg, A. B., Moss, A. S., Monti, D. A. & Alavi, A. Positron emission tomography in psychiatric disorders. Ann. N. Y. Acad. Sci. 1228, E13–25 (2011).
Wang, L. Y. et al. Performance evaluation of the Trans-PET® BioCaliburn® LH system: a large FOV small-animal PET system. Phys. Med. Biol. 60, 137–150 (2015).
Hitzemann, R. et al. Regional analysis of the brain transcriptome in mice bred for high and low methamphetamine consumption. Brain Sci. 9 (2019).
Zingg, B. et al. Neural networks of the mouse neocortex. Cell 156, 1096–1111 (2014).
Avey, D. et al. Single-cell RNA-seq uncovers a robust transcriptional response to morphine by glia. Cell Rep. 24, 3619–3629 (2018).
Zhu, L. et al. Chronic methamphetamine regulates the expression of MicroRNAs and putative target genes in the nucleus accumbens of mice. J. Neurosci. Res. 93, 1600–1610 (2015).
Gao, Q., Khan, S. & Zhang, L. Transcriptional profiling of mouse prefrontal cortex and nucleus accumbens under chronic jet lag. Gene Expression Omnibus http://identifiers.org/geo:GSE153540 (2020).
Gao, Q., Khan, S. & Zhang, L. Brain activity and transcriptional profiling in mice under chronic jet lag. figshare https://doi.org/10.6084/m9.figshare.c.5054273 (2020).
Acknowledgements
This work was supported by grants from the Natural Science Foundation of China (31930021 and 31671360) to LZ and China Postdoctoral Science Foundation (2018M632826) to QG. We would like to thank Drs. Na Gao and Weihua Chen at Huazhong University of Science and Techonology for helping to generate FastQC figures.
Author information
Authors and Affiliations
Contributions
L.Z. initiated the project and oversaw all aspects of the project. Q.G. and S.K. performed the experiments. Q.G. carried out the data analysis. Q.G. and L.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Gao, Q., Khan, S. & Zhang, L. Brain activity and transcriptional profiling in mice under chronic jet lag. Sci Data 7, 361 (2020). https://doi.org/10.1038/s41597-020-00709-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00709-6
