
Abstract
Genetic data are a crucial and exponentially growing resource across all biological sciences, yet curated databases are scarce. The widespread occurrence of sequence and (meta)data errors in public repositories calls for comprehensive improvements of curation protocols leading to robust research and downstream analyses. We collated and curated all available GenBank cytochrome-b sequences for amphibians, a benchmark marker in this globally declining vertebrate clade. The Amphibia’s Curated Database of Cytochrome-b (ACDC) consists of 36,514 sequences representing 2,309 species from 398 genera (median = 2 with 50% interquartile ranges of 1–7 species/genus). We updated the taxonomic identity of >4,800 sequences (ca. 13%) and found 2,359 (6%) conflicting sequences with 84% of the errors originating from taxonomic misidentifications. The database (accessible at https://doi.org/10.6084/m9.figshare.9944759) also includes an R script to replicate our study for other loci and taxonomic groups. We provide recommendations to improve genetic-data quality in public repositories and flag species for which there is a need for taxonomic refinement in the face of increased rate of amphibian extinctions in the Anthropocene.
Measurement(s) | DNA • mitochondrial_DNA • cytochrome b |
Technology Type(s) | digital curation • bioinformatics analysis |
Sample Characteristic - Organism | Amphibian |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12587744
Similar content being viewed by others

Background & Summary
Genetic data repositories are a key research component across scientific disciplines that rely on genetic sequences correctly assigned to a reference taxonomy. Although mistaken identity and composition of sequences within those repositories have long been acknowledged1,2,3,4,5, broad-scale data-quality evaluations remain scarce6,7,8 and rarely translate into improved databases. Therefore, the uncertainty of genetic data in global platforms such as GenBank3,9,10 represents a paramount obstacle for robust downstream analyses. Critically, quality-screening efforts can resolve misidentification of known, cryptic and undescribed taxa8,11, and inform the definition of reliable taxonomical units for management and biodiversity research12,13.
The widespread sequencing of, and access to, mitochondrial DNA (mtDNA) has boosted taxonomic studies via integrative taxonomy, barcoding, bioprospection, phylogenetics, phylogeography, population and conservation genetics, biogeography, macroecology, and paleoecology14,15,16. Available mtDNA data outcompetes nuclear DNA data in taxonomic coverage across the ‘Tree of Life’ mainly due to the popularity of 16 S, cytochrome-b (Cytb) and cytochrome oxidase 1 (Cox1) loci, while multiple sequences per species of those loci have proved crucial to define species limits17,18,19. While Cox1 was proposed as a universal barcode genetic marker20, GenBank’s Cytb records are currently more abundant than Cox1 for all five major vertebrate groups (Table 1).
Amphibians have the highest rate of newly discovered vertebrate species21 given intense taxonomic efforts11. These ectotherms are however the most threatened vertebrates on Earth22,23, with many species facing extinction owing to emerging and spreading diseases24,25, habitat loss26 and climate change27. Therefore, accurate phylogenetic identification11,28,29 remains critical for future research and conservation actions. Here, we present the Amphibia’s Curated Database of Cytochrome-b sequences (ACDC30, https://doi.org/10.6084/m9.figshare.9944759), a comprehensive and curated database of all amphibian Cytb sequences available in GenBank. We targeted Cytb because it is the most common genetic marker, with the broadest genus- and species-level taxonomic coverage, in the amphibian literature31,32.
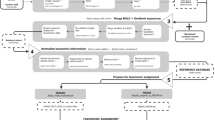
We created ACDC30 following a multi-step process implemented in a bioinformatic pipeline combining data retrieval from GenBank, local sequence alignments and quantification of genetic divergences (Fig. 1). On 01 February 2018, we retrieved a total of 39,202 Cytb sequences. Following curation (see Methods), ACDC contains 36,514 unique sequences representing 398 genera and 2,309 species (median = 2 species/genus with 50% interquartile ranges of [1,7]). For 1,363 species and 74 of the 75 amphibian families, there is more than one sequence available (Summary_statistics_ACDC.xlsx30) (median = 7 [3,22] species/family). ACDC represents 29% of the 7,963 currently known amphibian species covering most clades33. Despite the taxonomic accuracy of GenBank records seems to be accurate above the genus level34, our work demonstrates that the problematic issues mostly occur at the species level, and case-by-case assessments of taxonomic identity are necessary.

Workflow to collate and curate The Amphibia’s Curated Database of Cytochrome-b sequences (ACDC).
We identified 2,359 conflictive sequences (6% of the collated dataset) from 1,603 Anura, 743 Caudata, and 13 Gymnophiona records. These sequences suffered from wrong taxonomic assignments (>80%), contamination, introgression/hybridization, and submission/ sequencing errors (Fig. 2, Erroneous_sequences.xlsx30) and, as such, they qualify to be tagged as ‘UNVERIFIED’35 in GenBank. We updated the taxonomic identity of ca. 4,800 GenBank records (Taxonomic_corrections.xlsx30), and reverse-complemented reads from >1,000 sequences incorrectly uploaded as backward reads. We provide summary tables listing species/sequences with an uncertain taxonomic assignment (sp./ssp./cf./aff.; Uncertain_taxonomy_to_be_assessed.xlsx30) and potentially belonging to species complexes (Species_notes.xlsx30). These results suggest that several amphibian groups are in need of taxonomic revision. Lastly, we address general recommendations to improve data quality in public genetic repositories (Table 2) and append an R script30 to apply our data-curation protocol to other taxa and loci.

Frequency of error categories in amphibian Cytochrome-b sequences identified from GenBank sequences (01/02/2018). Those errors affect 6% (n = 2,359) of the sequences retrieved. Sequences identified due to incomplete lineage sorting are lumped in ‘Hybridization/Introgression’. Category definitions are explained in Erroneous_sequences.xlsx30.
Ideally, the research community would benefit from future sequencing efforts giving full taxonomic coverage to a selected sample of loci, which could in turn improve our understanding of amphibian biodiversity, evolution, ecology or conservation. mtDNA markers are still the best candidates to implement those efforts, as they are easy to amplify (even in poorly preserved samples), align and curate36. Taxonomic coverage of mtDNA can also be widened as a by-product of full-transcriptome and -genome assemblage, including long-read Next Generation Sequencing. In that respect, the development, integration, and expansion of quality-curated databases like ACDC should promote the generation of novel genomic data covering multiple specimens per species across the amphibian tree of life.
Methods
Workflow
Within the R environment37, on 01/02/2018, we used a key-word string to select and download all amphibian Cytb sequences from the GenBank’s website (www.ncbi.nlm.nih.gov/genbank, National Centre for Biotechnology Information) – see Steps 1–3 in the ACDCv1.0.R script30. We eliminated duplicates using GenBank labels ‘NC’, adjusted the nomenclature of each sequence to conform a genus_species_accession format (e.g., Bufo_bufo_AB123456), and exported all sequences as a single *.fasta file (Step 430). This includes single Cytb sequences, as well as mitochondrial genomes that contain this locus. All these sequences were then mapped against a reference mitochondrial genome (Xenopus tropicalis, AY789013), using the ‘high sensitivity’ option in Geneious® v11.038, and we extracted Cytb nucleotidic sequences (Fig. 1). Then, the nomenclature of all unique taxonomic identities was compared, confirmed and, if applicable, updated (Step 530) against the Amphibian Species of the World Database33.
We exported all mapped Cytb sequences in a *.fasta file from Geneious to the R environment. Therein, we performed ClustalW39 multiple sequence alignments for each species separately using the R package Bioconductor (Step 630). The resulting intraspecific alignments were imported back to Geneious as *.fasta files for batch-alignment through the MUSCLE algorithm (Fig. 1). The former step was mandatory because batch-MUSCLE alignments of multiple sequences (muscle function40 in Bioconductor) does not reorder sequences based on genetic similarity (A.T. Kalinka, pers. comm., 06/08/2018). Within Geneious, we visually resolved nucleotide gaps using the Vertebrate Mitochondrial Code41, and removed sequence ends with ambiguous nucleotides.
Taxonomic assessment and curation
We quantified accuracy on the assignation of sequences to species based on the genetic divergence (%) among sequences within species and genera and the identification of divergence outliers. We implemented three steps to detect sequencing and taxonomic errors based on pairwise-sequence alignments within each genus (Step 7; see Technical Validation). We used ‘uncorrected divergence’ as the genetic distance between every pair of sequences, using the seqinr package42. Firstly, we accepted sequences showing ≤3% divergence within multiple alignments across all sequences of the same species, and subset those with >3% divergence for further examination. Secondly, we also accepted sequences showing >3% divergence within a genus and subset those with ≤3% divergence for further examination. We caution that 3% is a reliable (conservative) divergence threshold for amphibian Cytb43,44,45,46 but should be re-estimated for other loci and taxonomical groups. Thirdly, for all potentially erroneous sequences, we assessed taxonomic and geographical veracity against (I) the data-source publication cited in GenBank, (II) the most recent papers dealing the taxon involved, (III) AmphibiaWeb (https://amphibiaweb.org) and (IV) the Amphibian Species of the World Database33 (Fig. 1). References and rationale used to separate erroneous from non-erroneous sequences are given for each sequence (Erroneous_sequences.xlsx30). We removed all erroneous sequences from ACDC and compiled all Amphibia Cytb sequences with uncertain taxonomy (aff./cf./sp./ssp.) (Uncertain_taxonomy_to_be_assessed.xlsx30). Lastly, the curation of genetic data is dependent on the number of available sequences per species and the taxonomic coverage per genus. Therefore, we included summary data for the ACDC database (Summary_statistics_ACDC.xlsx30) to flag species in need of more data and taxonomic resolution in online genetic repositories.
Lastly, our R script includes a routine to assess the Cytb region that maximizes species coverage and number of sequences (Supplementary Files 1 and 2). To do so, we first mapped all ACDC sequences to the Cytb of X. tropicalis (AY789013) using the ‘highest sensitivity’ option in Geneious, then counted non-missing bases for each position (Step 830).
Data Records
The curated database, all files as well as the associated R script are freely available on figshare30. The database consists of two compressed batches of *.fasta files of species with (I) 1 sequence (Species_with_One_Sequence.zip) and (II) > 1 sequences (Species_with_Multiple_Sequences.zip).
Technical Validation
We implemented a three-step sequence of filters to assess Cytb-sequence quality. (I) We retained sequences with complete binominal nomenclature. (II) We mapped all sequences against the Xenopus tropicalis mitochondrial genome (AY789013) and reverse-complemented sequences incorrectly submitted in backward-read format (>1,000). (III) We visually scanned sequence alignments for sequencing errors, whereby non-amino acid gaps (≠3) were filled or replaced by ‘N’ in the absence or presence of diversity at the base in question, respectively.
Code availability
The R script used to collate and curate the Amphibia Cytb database is available at figshare (ACDCv1.0.R30).
References
Brunak, S., Engelbrecht, J. & Knudsen, S. Neural network detects errors in the assignment of mRNA splice sites. Nucleic Acids Res 18, 4797–4801 (1990).
Harris, D. Can you bank on GenBank? Trends Ecol. Evol. 18, 317–319 (2003).
Wesche, P. L., Gaffney, D. J. & Keightley, P. D. DNA sequence error rates in Genbank records estimated using the mouse genome as a reference. DNA Seq. 15, 362–364 (2004).
Buhay, J. E. “COI-like” Sequences are becoming problematic in molecular systematic and DNA barcoding studies. J. Crustac. Biol 29, 96–110 (2009).
Alkan, C., Sajjadian, S. & Eichler, E. E. Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65 (2011).
Machida, R. J., Leray, M., Ho, S.-L. & Knowlton, N. Data Descriptor: Metazoan mitochondrial gene sequence reference dataset for taxonomic assignment of environmental samples. Sci. Data 4, 170027 (2017).
Heller, P., Casaletto, J., Ruiz, G. & Geller, J. Data Descriptor: A database of metazoan cytochrome c oxidase subunit I gene sequences derived from GenBank with CO-ARBitrator. Sci. Data 5, 180156 (2018).
Li, X. et al. Detection of potential problematic Cytb gene sequences of fishes in GenBank. Front. Genet 9, 30 (2018).
Prada, C. F. & Boore, J. L. Gene annotation errors are common in the mammalian mitochondrial genomes database. BMC Genomics 20, 73 (2019).
Ross, H. A. & Murugan, S. Using phylogenetic analyses and reference datasets to validate the species identities of cetacean sequences in GenBank. Mol. Phylogenetics Evol 40, 866–871 (2006).
Vieites, D. R. et al. Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proc. Natl. Acad. Sci. 16, 8267–8272 (2009).
Shen, Y.-Y., Chen, X. & Murphy, R. W. Assessing DNA barcoding as a tool for species identification and data quality control. PLoS ONE 8, e57125 (2013).
Morin, P. A. et al. Applied conservation genetics and the need for quality control and reporting of genetic data used in fisheries and wildlife management. J. Hered. 101, 1–10 (2010).
Gershoni, M., Templeton, A. R. & Mishmar, D. Mitochondrial bioenergetics as a major motive force of speciation. BioEssays 31, 642–650 (2009).
Toews, D. P. L. & Brelsford, A. The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol 21, 3907–3930 (2012).
Ballard, J. W. O. & Pichaud, N. Mitochondrial DNA: More than an evolutionary bystander. Funct. Ecol. 28, 218–231 (2013).
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H. & Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. 101, 14812–14817 (2004).
Čandek, K. & Kuntner, M. DNA barcoding gap: Reliable species identification over morphological and geographical scales. Mol. Ecol. 15, 268–277 (2014).
Liu, J. et al. Multilocus DNA barcoding – Species Identification with multilocus data. Sci. Rep. 7, https://doi.org/10.1038/s41598-017-16920-2 (2017).
Herbert, P. D., Cywinska, A., Ball, S. L. & de Waard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 270, 313–321 (2003).
Köhler, J. et al. New amphibians and global conservation: A boost in species discoveries in a highly endangered vertebrate group. BioSience 55, 693–696 (2005).
Stuart, S. N. et al. Status and trends of amphibian declines and extinctions worldwide. Science 306, 1783–1786 (2004).
IUCN. The IUCN Red List of Threatened Species. Version 2018-2 (2019).
Martel, A. et al. Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346, 630–631 (2014).
Lips, K. R. Overview of chytrid emergence and impacts on amphibians. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150465 (2016).
Cushman, S. A. Effects of habitat loss and fragmentation on amphibians: A review and prospectus. Biol. Conserv. 128, 231–240 (2006).
Winter, M. et al. Patterns and biases in climate change research on amphibians and reptiles: A systematic review. R. Soc. Open Sci. 3, 160158 (2016).
Liu, Z. et al. Prevalence of cryptic species in morphologically uniform taxa – Fast speciation and evolutionary radiation in Asian frogs. Mol. Phylogenetics Evol 127, 723–731 (2018).
Funk, W. C., Caminer, M. & Ron, S. R. High levels of cryptic species diversity uncovered in Amazonian frogs. Proc. R. Soc. Lond. B Biol. Sci. 279, 1806–1814 (2011).
van den Burg, M. P., Herrando-Pérez, S. & Vieites, D. R. ACDC, a curated database of amphibian cytochrome-b sequences. figshare https://doi.org/10.6084/m9.figshare.9944759.v2 (2020).
Grant, T. et al. Phylogenetic systematics of dart-poison frogs and their relatives (Amphibia: Athesphatanura: Dendrobatidae). Bull. Am. Mus. Nat. Hist. 121, 1–263 (2006).
Pyron, R. A. & Wiens, J. J. A large-scale phylogeny of Amphibia including over 2800 species, and a revised classification of extant frogs, salamanders, and caecilians. Mol. Phylogenetics Evol 61, 543–583 (2011).
Frost, D. R. Amphibian Species of the World: an Online Reference, Version 6.0. American Museum of Natural History http://research.amnh.org/herpetology/amphibia/index.html (2018).
Layer, M. et al. GenBank is a reliable resource for 21st century biodiversity research. Proc. Natl. Acad. Sci. 116, 22641–22656 (2019).
Benson, D. A. et al. GenBank. Nucleic Acids Res 40, 48–53 (2012).
Harrison, R. G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 4, 6–11 (1989).
R v.3.6.2. (R Core Team, 2018).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Bodenhofer, U., Bonatesta, E., Horejs-Kainrath, C. & Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 31, 3997–3999 (2015).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Elzanowski, A. & Ostell, J. The Genetic Codes, https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi?chapter=tgencodes#SG2 (2019).
Charif, D. & Lobry, J. R. In Structural approaches to sequence evolution: Molecules, networks, populations Vol. 1 (ed. Bastolla, U. et al) Ch. 10 (Springer Verlag, 2007).
Vences, M., Thomas, M., Van Der Meijden, A., Chiari, Y. & Vieites, D. R. Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Front. Zool. 2, 5 (2005).
Vences, M., Thomas, M., Bonett, R. M. & Vieites, D. R. Deciphering amphibian diversity through DNA barcoding: chances and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci 360, 1859–1868 (2005).
Johns, G. J. & Avise, J. C. A comparative summary of genetic distances in the vertebrate from the mitochondrial cytochrome b gene. Mol. Biol. Evol. 15, 1481–1490 (1998).
Smith, M. A., Poyarkov, N. A. Jr. & Hebert, D. N. CO1 DNA barcoding amphibians: take the chance, meet the challenge. Mol. Ecol. Resour 8, 235–246 (2008).
Acknowledgements
We are grateful to Angus and Malcolm Young, Brian Johnson, Cliff Williams, and Phill Rudd for their contribution to a productive and relaxing working atmosphere. This work was supported by the Ministerio de Ciencia y Competitividad grant CGL2017-89898-R (AEI/FEDER, EU) grant to DRV.
Author information
Authors and Affiliations
Contributions
D.R.V. designed the study. M.P.v.d.B. curated and filtered the data, and wrote the first draft. S.H.P. wrote the R script. All authors contributed to the Data Descriptor and contributed to revisions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
van den Burg, M.P., Herrando-Pérez, S. & Vieites, D.R. ACDC, a global database of amphibian cytochrome-b sequences using reproducible curation for GenBank records. Sci Data 7, 268 (2020). https://doi.org/10.1038/s41597-020-00598-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-00598-9
