
Abstract
In the recent years, the improvement of software and hardware performance has made biomolecular simulations a mature tool for the study of biological processes. Simulation length and the size and complexity of the analyzed systems make simulations both complementary and compatible with other bioinformatics disciplines. However, the characteristics of the software packages used for simulation have prevented the adoption of the technologies accepted in other bioinformatics fields like automated deployment systems, workflow orchestration, or the use of software containers. We present here a comprehensive exercise to bring biomolecular simulations to the “bioinformatics way of working”. The exercise has led to the development of the BioExcel Building Blocks (BioBB) library. BioBB’s are built as Python wrappers to provide an interoperable architecture. BioBB’s have been integrated in a chain of usual software management tools to generate data ontologies, documentation, installation packages, software containers and ways of integration with workflow managers, that make them usable in most computational environments.
Similar content being viewed by others

Introduction
Biomolecular simulations have attained in the last years a level of maturity that allows to use them as “computational microscopes” to gain insight in biological processes. Atomistic simulations extend now to the μs range, approaching the time range of biological processes1,2. Coarse-grained simulations can go even further, in the length of simulations, and the size of the systems that can be analysed3,4,5,6. The traditional scope of simulations has overpassed the single protein or small nucleic acid systems to deal with relevant multiprotein and protein-nucleic acid complexes, nucleosomes, long segments of RNA, sections of chromatin or even full chromosomes5. This scenario envisions now a clear bridge between biomolecular simulations and genomics. Multiscale approaches can now bring together, for instance, Chip-seq data with simulation of protein-DNA complexes, or HiC or oligopaint FISH experiments with large scale simulations of chromatin fibers5. However, the type of tools, and the way they are used differ between genomics and biomolecular simulations. Simulations have been traditionally based on a reduced number of well optimized codes run in HPC systems, where they indeed occupy a large amount of resources (over 60 M CPU-hours of BSC’s MareNostrum supercomputer were dedicated to biomolecular simulations in 2018). On the other hand, traditional bioinformatics uses many competing tools usually orchestrated in complex workflows. Considering data, genomics mobilizes indeed the major amount of it, however, the storage of a typical μs-range trajectory on a mid-sized system requires already some hundreds of GB like a human whole genome obtained by Next-Generation Sequencing (NGS).
Workflow orchestration is a well-accepted concept in bioinformatics. No single, universal, solution exists, and the number of available frameworks to build and run workflows is large (https://github.com/common-workflow-language/common-workflow-language/wiki/Existing-Workflow-systems). Initiatives in the past like myGrid7 and BioMoby8, or more recent initiatives like CWL9, or WDL (https://software.broadinstitute.org/wdl/), have attempted to define an interoperable ecosystem to run bioinformatics tools, web-services and the workflows made out of them. Managers like Taverna10, Rabix11, Cromwell (https://cromwell.readthedocs.io/en/stable/), KNIME12 or repositories like myExperiment13 allow to execute or store workflow definitions for further re-usage. In this context, the ELIXIR (http://elixir-europe.org) organization is working to put in place recommendations to organize such ecosystem. At the level of registration, bio.tools14 and Fairsharing15 provide repositories for tools and standards. Specification languages like openAPI (https://www.openapis.org/), and CWL9 are being recommended to document APIs and workflows, respectively. In terms of workflow execution, Galaxy16 appears as the most popular framework, although other managers are also commonly used (e.g. Nextflow17, PyCOMPSs18, Snakemake)19. To formalize this scenario, the FAIR principles20, initially presented to improve the quality of scientific data, are now being extended to research software. The key requirements for that (registries, standards, software managers and open repositories) are already available. Several organizations including the Software Sustainability Institute (https://www.software.ac.uk/), Research Software Engineers’ associations, or ELIXIR itself are participating actively in the discussion.
Bioinformatics initiatives have little application to the simulation world. Simulations themselves are run in HPC systems in highly optimized environments. Most of the work, like setting up the simulation, a key step to assure the quality of the results21,22,23,24, and the analysis of trajectories, is done almost manually. Modelers use in-house scripts, typically based on the software included in the simulation packages. In this situation, researchers usually limit themselves to a single package for all steps: setup, simulation, and analysis. Therefore, since the possibility of complementing software functionalities across packages is limited, developers should provide complete sets, re-implementing what other packages provide already. Additionally, since data formats are also diverse, data conversion modules proliferate, what in turn raises the question of which combinations of tools (although theoretically compatible) would give correct scientific answers.
Efforts to automate simulation setup and analysis do exist. Several graphical interfaces have been designed to ease the interaction with specific simulation packages25,26,27,28,29,30. These tools are especially useful for non-experts as they simplify the learning process. However, these utilities are still linked to specific simulation packages. One of the attempts, by our group, was MDWeb31. This was the first approach to offer a unified workbench allowing to setup a protein system for atomistic molecular dynamics simulation, able to work for GROMACS32, NAMD33, and Amber34, three of the most popular simulation packages. Remarkably, MDWeb is powered internally by a series of web services built within the BioMoby framework and uses a common ontology of data types for the three simulation packages (http://mmb.irbbarcelona.org/MDWeb2/help.php?id=ontology). In this sense, this attempt, still in use with over 3,000 registered users, was rather unique. MDWeb was extended to the nucleic acids world with a nucleic-acids specific analysis portal, NAFlex35. At the large-scale end, systems have been designed to manage large scale simulation projects. Copernicus36 combines peer-to-peer communication strategies with a simulation specific workflow management system, able to control large simulation sets in a distributed computational network. The iBIOMES project37,38 reported an infrastructure to manage and share distributed simulation data, based in the iRODS framework (https://irods.org/). iBioMES has been used recently to manage nucleosome simulation data39, in a clear example for the growing overlap between simulation and genomics. Some simulation databases have also been built. Dynameomics40, centered in analysing protein folding and stability, MoDEL22 offering an initial attempt of covering a significant sample of known protein structures, and BigNASim23, specialized in Nucleic Acids. Remarkably, MoDEL and BigNASim provided ontologies for representing simulation data (https://mmb.irbbarcelona.org/BIGNASim/help.php?id=onto).
Even though a large set of tools are normally combined, the concept of workflow, as understood in general bioinformatics, is of limited usage. As said, most systems are setup and analyzed using in-house scripts. Recently, the BioExcel Center of Excellence (http://bioexcel.eu) has taken the objective of pushing the concept and usage of workflows into the biomolecular research field. In this work, we present a comprehensive exercise joining ELIXIR’s recommendations and services, FAIR principles, and biomolecular simulations. We have selected the automatic setup for molecular dynamics simulations of a protein system including sequence variants, as case for demonstration. The aim of the exercise is to assess the feasibility of working according the FAIR principles and ELIXIR’s recommendations in a field that is considered out of the scope of common bioinformatics. We will present a fully interoperable software library (the BioExcel Building Blocks, BioBBs) based mainly on (but not limited to) GROMACS32 software components. For the deployment of BioBBs, we have leveraged existing platforms and services commonly used in bioinformatics, like BioConda41, BioContainers42 or Galaxy16. Workflows built using components of such library have been executed in several complementary computational environments, including personal desktops, virtualized systems, public e-infrastructures, and HPC systems. Besides, the components are documented using CWL and openAPI, what opens the possibility of run them in CWL complaint workflow managers.
Results and Discussion
Moving toward FAIR principles
FAIR principles20 were defined with the aim of improving the quality of bioinformatics data repositories. Main principles include (1) Findability: Data should be findable, i.e. identified by permanent identifiers and included in searchable registries; (2) Accessibility: Data should be stored in permanent repositories and accessible in a machine readable form, (3) Interoperability: Data should use well-documented formats and standards to allow to interoperate with complementary datasets; and (4) Reusability: Documentation about the conditions and limitations of data reusability should be provided. Adherence to these principles has become part of the best-practices in bioinformatics data management and begins to be generally understood and accepted by the research community. They cannot be applied blindly to research software, but the general guidelines can be adapted.
Findability
A primary requirement for findability in the case of software is the availability of a software registry. Traditional software repositories like GitHub (https://github.com), are suitable for such usage although they are not usually seen as data resources, and the amount of available scientific metadata is limited. To overcome this limitation, registries with different degrees of acceptance exist (https://www.genscript.com/tools.html; https://omictools.com/; https://www.fda.gov/ScienceResearch/BioinformaticsTools/default.htm). ELIXIR has pushed its own tools registry (bio.tools)14. It includes a large set of metadata that allows to search for tools according to their scientific utility, and provides extended metadata regarding publications, documentation and support. It is linked to ELIXIR’s software benchmarking platform, openEBench (https://openebench.bsc.es), which in turn provides data for technical and scientific quality assessment of bioinformatics applications. One of the most remarkable features of bio.tools is the use of an extended ontology (EDAM43) for annotation. EDAM annotations allow to classify tools according the type of data they consume or produce and provides a controlled vocabulary to define their precise functionality. This information has been used to derive tools’ annotation for CWL, or Galaxy44,45 automatically. Unfortunately, ontology terms for structural bioinformatics, in general, and biomolecular simulation specifically, were scarce in EDAM. The generation of ontologies on simulation have been attempted in the past22,23, but such ontologies have been seldom used outside the projects that generated them. However, the interest for addressing simulation data management has increased recently24.
The first step of this exercise was to essay the registration in bio.tools of tools required for setup and analysis of a protein simulation. From this assay, several missing data types, file formats, and functionalities were collected (see Supplementary Table S1). We have taken the experience in MDWeb31, MoDEL22, and BigNASim23 ontologies to fill the gaps in EDAM. The additions included setup, simulation and analysis operations, specific data types like system topology, trajectories, or principal components, and file formats covering the most popular simulation codes (Supplementary Table S1). These new terms have been already included in EDAM v1.22 (https://raw.githubusercontent.com/edamontology/edamontology/master/EDAM_dev.owl) and will be available for tools annotation in short. More than thirty simulation related tools, besides the BioBB library components have been registered in bio.tools. To provide an additional means for findability, a BioSchemas – based specification (http://bioschemas.org/) has been included in the appropriate places of BioBB’s documentation.
A software architecture for interoperability
The recipe for full tool interoperability is theoretically simple: the use of a common, universal, data model. Past attempts like myGrid7 and BioMoby8 put foundations to this concept, by building a community-based data ontology and suggesting tool developers to stick to it when generating new tools. However, this attempt was not successful. The community-based approach made difficult to keep control on the evolution of the ontology. Similarly, in Galaxy servers, for instance, system administrators may add ad-hoc types and formats, hence contributing to make the scenario even more complex. In summary, attempting to generate a common data model for bioinformatics remains as a hard issue. Fortunately, when we focus on specific fields (NGS, array analysis, etc.), the options are limited, and de-facto standards do exist (bam, vcf, gff file formats in NGS analysis, for instance). Similarly, in biomolecular simulation the limited number of software packages makes the scenario less complicated. In any case, however, tool interoperability is an issue; a large set of operations in bioinformatics are, in fact, format conversions, and there is no security that an input data file is compatible with a given tool, even though that the format is the correct one.
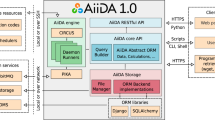
In this exercise, we have defined a specific software architecture to contribute to the interoperability (Fig. 1). We use simple wrappers, written in Python, to encapsulate software components. Wrappers are organized in layers. The inner layer corresponds to the original tool, unaltered. Command-line tools, web services, software containers, or even remote calls to HPC systems, can be included here. A second one, the compatibility layer, provides the module with a well-defined interface for input, output, configuration, and provenance. It performs internally the necessary format conversions at input and output and launches the tool. This interface can be fully documented and specified using accepted standards like openAPI or CWL and can remain stable even when the associated tool needs to be updated. These two-layer wrappers can be already integrated in scripts as Python modules or executed as standalone command-line tools. A third layer, the adaptor, may be required for the integration in execution engines or e-infrastructures. BioBBs adaptors for Galaxy, PyCOMPSs, and CWL compliant managers are provided. Such adaptors can be used as templates to extend the usability of the library to other environments.

BioExcel building block architecture. BioBB’s structure split in three main layers: The inner layer corresponds to the original tool unaltered, the second one, the Python compatibility layer provides a standardized interface, the third one, the outer workflow manager adaptation layer translates the Python standard interface to each specific WF manager.
This architecture, even though it does not provide a common data model, do provide a uniform and stable interface, with enough information to plug the components into interoperable workflows (see below). Besides, any updates in the inner software tool would require only to update the wrapper, maintaining compatibility with previous versions, workflows, and with the chosen deployment options. Table 1 shows the present list of BioBBs with indication of their functionalities and associated tools.
Providing accessibility and enabling (re)usability
In the case of tools, the accessibility requirement is even stricter than for data: software not only should be accessible, it needs to be installed and executed. Different execution scenarios should be considered in the case of biomolecular simulations. They include personal workstations, used mainly for setup and analysis, or HPC systems where simulations are usually obtained. To address this principle, BioBB’s use several deployment possibilities. Figure 2 shows a global information flow, and Online-only Table 1 summarizes the URLs corresponding to the different BioBB deployment alternatives. The main software repository used is available on Github. Information embedded in the code allows to generate (1) documentation using the ReadTheDocs platform (https://readthedocs.org/), (2) a JSON schema for library specification using openAPI, and (3) a reference CWL specification. To ease the deployment in a complete set of environments we have put together several packaging systems and services (Fig. 2). From the code deposited in Github, BioBBs have been uploaded to the Python Packaging Index Pypi (https://pypi.org/). Also, BioConda41 packages have been prepared. These will allow to handle software dependencies in a transparent way, including the installation of the embedded tools. Considering only these two options, the package would be already available for installation where command-line is the main execution procedure, like personal workstations, clusters, virtual machines, or HPC. Installation can be done both as system-wide Python packages or using Python virtual environments. This kind of installation is illustrated by the execution of the lysozyme test (see below) in a Jupyter Notebook (https://jupyter.org/). Following from BioConda packages, and due to its integration with the BioContainers project42, Docker containers are automatically generated and deposited in the quay.io repository. Offered Docker containers provide functionality for either individual packages to be integrated in more complex layouts, or complete workflows. Docker containers, in turn, are converted to Singularity containers that can be used in security demanding environments like HPC. Containers allow the non-expert user to deploy the software easily. For instance, Docker containers have been used to deploy BioBBs in a test Galaxy installation (http://dev.usegalaxy.es). BioBBs, encapsulated as Virtual Machines, are also available on BioExcel cloud portal (https://bioexcel.ebi.ac.uk), and EGI’s appDB (https://appdb.egi.eu). Table 2 summarizes the recommended installation and execution options for the environments tested in the project.

Recommended distribution and deployment flow of the BioBBs. Distribution and packaging tools used to facilitate BioBB’s installation and execution in a wide range of platforms: HPC, Cloud computing, user workstations and even browser interfaces.
BioBBs are fully open source, distributed under the Apache-2 license. Wrapped applications have their own licensing schemes, but for the library provided at present only open source software has been included.
Testing BioBBs in several environments. Setup for simulation for protein variants workflow
To test the feasibility of the software architecture, we have chosen a well-known procedure, the setup in standard conditions for molecular dynamics simulations of a protein system with sequence variants. We have used two biological systems: Lysozyme (PDB id 1AKI)46, and Pyruvate kinase (PDB id 2VGB)47. Lysozyme is a small protein (129 res), which structure is available at a high resolution. The second system, Pyruvate kinase is a 200 kDa homo-tetramer, meaning a ~400,000 atom system after setup. Pyruvate kinase is a well-studied system with relevance in the understanding of allosteric regulation, but also of biomedical interest: more than 200 sequence variants related to pathogenic effects have been reported48. The test-cases consisted in a standard setup for NPT simulation with explicit solvent of several selected variants, followed with 5 ns long simulations, and a simple RMSd comparative analysis (see Method section). Supplementary Figs S1 and S2 show a schema of the simulation setup workflow as rendered by CWL viewer (https://view.commonwl.org) and Galaxy respectively. We have tested (1) the feasibility of running the workflow (including software installation, and workflow execution) in a variety of computational environments (Lysozyme test) and (2) its scalability on HPC systems (Pyruvate kinase test). Supplementary Table S3 shows a summary of the architectures and the executions performed. Execution times are shown just for illustration purposes and are totally dependent on the hardware used. Since most of the execution time corresponds to the simulation phases, no significant overhead in using the different execution approaches was detected. Parallelization has been carried out at different levels. PyCOMPSs has been used to deal with simulations of different protein variants, at a ratio of 1 variant per process. GROMACS parallelization schemes (OpenMP for intra-node parallelization and MPI when several nodes were involved) were used in the simulation phase. Linear scaling has been observed in all cases (note the similar wall-clock times between the two extreme executions made at BSC’s MareNostrum, ranging from 2 variants, 384 cores, to 200 variants, 38,400 cores).
Conclusions
Biomolecular simulations are seldom considered as part of the field known as bioinformatics, even structural bioinformatics. Reasons for that come not only from the use of a different kind of tools and computational resources, but also from the traditional lack of applicability of simulation results to day-to-day biology. In the recent years, simulation has attained a significant level of maturity, and simulation results are now compatible with biologically relevant systems and time scales. Biomolecular simulations are already tackling questions that can be relevant for genomics, or transcriptomics. However, the isolation of biomolecular simulations in the context of bioinformatics has prevented the adoption by this community of normal software trends in bioinformatics, like automatic software deployment or the use of workflow managers. We have presented here the exercise of treating biomolecular simulations as normal bioinformatics operations. To this end, we have decorated standard simulation operations with a series of concepts and procedures, like an initial adherence to FAIR principles, the usage and documentation of workflows and stable interfaces, and the availability of a variety of deployment options, that are becoming routine in bioinformatics. FAIR principles for software have not yet been defined in the way as they exist for data. The exercise has led to an approach to the selection of software features (registration, methods of installations and deployment, documentation, licenses) that can be considered as an initial approach to them. The main outcome of the exercise is a complete software library (the BioBBs) that can be installed, deployed, and used as traditional bioinformatics applications, but provides a set of operations related to biomolecular simulations. BioBBs have been incorporated to the bioinformatics ecosystem: (1) The necessary new terms have been added to EDAM ontology, and tools included in the bio.tools registry. Bio.tools would provide a permanent identifier for them and the required metadata to assure their findability; (2) Interoperability has been addressed by the design of BioBBs architecture, but also through the use of recommended standards for specification (OpenAPI, CWL); and (3) Accessibility and usability have been addressed by using the set of well-known utilities, like Pipy, BioConda, BioContainers, or Galaxy, allowing the deployment and test of the library in a variety of alternative environments, from personal workstations to HPC.
BioBBs align with a variety of software that focus in similar functionality, however it opens the integration of biomolecular simulation operations into a more general bioinformatics landscape using similar, and compatible, software management procedures.
Methods
Atomistic simulations
Lysozyme test
Simulation of two sequence variants (Val2Tyr, and Val2Ala) of chicken Lysozyme (PDB code 1AKI)46 were prepared as follows. Protein structure was obtained from the Protein Data Bank49. Amino acid side chains were modified as appropriate using the biobb_model package. Hydrogen atoms were added to the structure using standard ionization at pH 7.0. Protein was placed in a Cubic box of explicit water solvent (SPC/E water model)50 with the appropriate size to allow 1 nm from the outermost protein atom. Periodic Boundary Conditions were applied. Cl− and Na+ ions were added to reach an ion concentration of 0.05 M and neutralize the system. Simulations were run using GROMACS 2018, and the Amber99sb-ILDN forcefield51. Temperature was maintained at 300 K and pressure to 1 atm. Setup was completed by 5,000 steps of steepest-descent energy minimization, followed by a 10 ps-long NVT equilibration, and a 10 ps-long NPT equilibration runs with a restriction of 1,000 kJ/mol.nm2 put on heavy atoms. Production phase for the test consisted in 5 ns of unbiased NPT simulation at 2 fs time step. The LINCS algorithm52 was used to keep covalent bonds at their equilibrium distances. Simulation setup and equilibration were done using components of the biobb_md package.
Pyruvate kinase test
200 sequence variants for Human erythrocyte Pyruvate kinase (PDB code 2VGB)47 were obtained from UniprotKB53 (biobb_io package). Protein structure was obtained from the Protein Data Bank49. All non-protein components of the structure were removed, and protein variants were prepared by modification of the appropriate amino acid side chains using biobb_model package. Hydrogen atom were added considering standard ionization states at pH 7.0. Simulation was done in a truncated octahedron box placed at a distance of 1.5 nm from the outermost atom of the protein, using TIP3P water molecules54, and using Periodic Boundary Conditions. Ions Cl− and Na+ were added to reach an ion concentration of 0.05 M and neutralize the system. The Particle mesh Ewald method55 was used to calculate electrostatic and Van der Waals interactions, with 0.12 nm of FF grid spacing and a cut-off distance of 1 nm for both Coulomb and Lennard-Jones interactions. The LINCS algorithm52 was used to keep covalent bonds at their equilibrium distances. Simulations were run using GROMACS 2018, and the Amber99sb-ILDN forcefield51. Temperature was maintained constant at 300 K (except in gradual heating), in two separate baths for the protein and non-protein groups, with the V-rescale thermostat56 and a coupling constant of 0.1 ps. Pressure was isotropically maintained at 1 bar in NPT ensembles through Parrinello-Rahman coupling57 with a constant of 1 ps, and applying a scaling of the center of mass of the reference coordinates with the scaling matrix. Given the size and complexity of the system, the Pyruvate kinase equilibration was performed with a more extended procedure: Setup was completed with two 5,000 steps energy minimizations, the first with a restrained potential of 500 kJ.mol−1.nm−2 on all heavy atoms except those in the side chain of the mutated residue, and the second with all heavy atoms restrained. Systems were then equilibrated with the following steps: (1) 100 ps of gradual heating from 0 to 300 K with 1,000 kJ.mol−1.nm−2 of restrained potential in heavy atoms except for mutated side chains, (2) four 20 ps steps of equilibration with descending restrain force constants in the same atoms (from 1,000 to 300 kJ.mol−1.nm−2), (3) two 10 ps steps of NPT equilibration with restraints in all backbone atoms (200 and 100 kJ.mol−1.nm−2 respectively) and (4) a 100 ps NPT equilibration without restraints. After equilibration, we ran 5 ns of unbiased NPT simulation. Simulation setup and equilibration were done using components of the biobb_md package.
Computational systems used
Systems used on Lysozyme test were: Workstation: ThinkStation E30 (LENOVO). Operating system: Linux Ubuntu 18.04. 8 CPU Intel(R) Xeon(R) CPU E31230 @ 3.20 GHz (1 socket, 4 cores/socket, 2 threads/core). 16 GB of RAM. Virtual Machine: 12 CPU QEMU Virtual CPU version 2.5+. 24 GB of RAM. Galaxy: 2 CPU QEMU Virtual CPU version 2.5+. Pyruvate kinase test was performed on BSC’s MareNostrum supercomputer using from 2 to 800 nodes of 2x Intel Xeon Platinum 8160 24C at 2.1 GHz, 12 × 8 GB of RAM. Largest test used 4 nodes per simulation with a total of 38,400 cores.
Code Availability
BioBB’s source code is available at GitHub. URLs for the code and documentation repositories and the alternative installation and execution options are summarized in Online-only Table 1.
References
Hospital, A. & Gelpi, J. L. High-throughput molecular dynamics simulations: toward a dynamic view of macromolecular structure. Wiley Interdisciplinary Reviews-Computational Molecular Science 3, 364–377 (2013).
Orozco, M. A theoretical view of protein dynamics. Chem. Soc. Rev. 43, 5051–5066 (2014).
Ayton, G. S., Noid, W. G. & Voth, G. A. Multiscale modeling of biomolecular systems: in serial and in parallel. Current Opinion in Structural Biology 17, 192–198 (2007).
Emperador, A., Carrillo, O., Rueda, M. & Orozco, M. Exploring the suitability of coarse-grained techniques for the representation of protein dynamics. Biophysical Journal 95, 2127–2138 (2008).
Dans, P. D., Walther, J., Gómez, H. & Orozco, M. Multiscale simulation of DNA. Curr Opin Struct Biol 37, 29–45 (2016).
Dans, P. D. et al. Modeling, Simulations, and Bioinformatics at the Service of RNA Structure. Chem 5, 51–73 (2019).
Stevens, R. D., Robinson, A. J. & Goble, C. A. myGrid: personalised bioinformatics on the information grid. Bioinformatics 19(Suppl 1), i302–4 (2003).
Wilkinson, M. D. et al. Interoperability with Moby 1.0–it’s better than sharing your toothbrush! Brief Bioinform 9, 220–231 (2008).
Amstutz, P. et al. Common Workflow Language, v1.0. figshare. https://doi.org/10.6084/M9.FIGSHARE.3115156.V2 (2016).
Wolstencroft, K. et al. The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic acids research 41, W557–W561 (2013).
Kaushik, G. et al. RABIX: An Open-Source Workflow Executor Supporting Recomputability and Interoperability of Workflow Descriptions. Pacific Symposium on Biocomputing 22, 154–165 (2016).
Beisken, S. et al. KNIME-CDK: Workflow-driven cheminformatics. BMC bioinformatics 14, 257–257 (2013).
Goble, C. A. et al. myExperiment: a repository and social network for the sharing of bioinformatics workflows. Nucleic acids research 38, W677–W682 (2010).
Ison, J. et al. Tools and data services registry: a community effort to document bioinformatics resources. Nucleic acids research 44, D38–D47 (2016).
McQuilton, P. et al. BioSharing: curated and crowd-sourced metadata standards, databases and data policies in the life sciences. Database: the journal of biological databases and curation 2016, baw075 (2016).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic acids research 46, W537–W544 (2018).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nature Biotechnology 35, 316 (2017).
Tejedor, E. et al. PyCOMPSs: Parallel computational workflows in Python. The International Journal of High Performance Computing Applications 31, 66–82 (2015).
Köster, J. & Rahmann, S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522 (2012).
Wilkinson, M. D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data 3, 16008 (2016).
Rueda, M. et al. A consensus view of protein dynamics. Proc Natl Acad Sci USA 104, 796–801 (2007).
Meyer, T. et al. MoDEL (Molecular Dynamics Extended Library): a database of atomistic molecular dynamics trajectories. Structure 18, 1399–1409 (2010).
Hospital, A. et al. BIGNASim: a NoSQL database structure and analysis portal for nucleic acids simulation data. Nucleic Acids Res 44, D272–278 (2016).
Elofsson, A. et al. Ten simple rules on how to create open access and reproducible molecular simulations of biological systems. PLoS computational biology 15, e1006649–e1006649 (2019).
Kota, P. GUIMACS - a Java based front end for GROMACS. In Silico Biol 7, 95–99 (2007).
Miller, B. T. et al. CHARMMing: a new, flexible web portal for CHARMM. Journal of chemical information and modeling 48, 1920–1929 (2008).
Jo, S. et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. Journal of computational chemistry 38, 1114–1124 (2017).
Sellis, D., Vlachakis, D. & Vlassi, M. Gromita: a fully integrated graphical user interface to gromacs 4. Bioinformatics and biology insights 3, 99–102 (2009).
Roopra, S., Knapp, B., Omasits, U. & Schreiner, W. jSimMacs for GROMACS: A Java Application for Advanced Molecular Dynamics Simulations with Remote Access Capability. J. Chem. Inf. Model. 49, 2412–2417 (2009).
Ribeiro, J. V. et al. QwikMD - Integrative Molecular Dynamics Toolkit for Novices and Experts. Scientific reports 6, 26536–26536 (2016).
Hospital, A. et al. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics 28, 1278–1279 (2012).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England) 29, 845–854 (2013).
Nelson, M. T. et al. NAMD: a Parallel, Object-Oriented Molecular Dynamics Program. The International Journal of Supercomputer Applications and High Performance Computing 10, 251–268 (1996).
Pearlman, D. A. et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Computer Physics Communications 91, 1–41 (1995).
Hospital, A. et al. NAFlex: a web server for the study of nucleic acid flexibility. Nucleic Acids Res 41, W47–55 (2013).
Pronk, S. et al. Molecular Simulation Workflows as Parallel Algorithms: The Execution Engine of Copernicus, a Distributed High-Performance Computing Platform. J. Chem. Theory Comput. 11, 2600–2608 (2015).
Thibault, J. C., Facelli, J. C. & Cheatham, T. E. 3rd. iBIOMES: managing and sharing biomolecular simulation data in a distributed environment. J Chem Inf Model 53, 726–736 (2013).
Thibault, J. C., Cheatham, T. E. 3rd. & Facelli, J. C. iBIOMES Lite: summarizing biomolecular simulation data in limited settings. J Chem Inf Model 54, 1810–1819 (2014).
Sun, R., Li, Z. & Bishop, T. C. TMB-iBIOMES: An iBIOMES-Lite Database of Nucleosome Trajectories and Meta-Analysis. Preprint at, https://doi.org/10.26434/chemrxiv.7793939.v1 (2019).
van der Kamp, M. W. et al. Dynameomics: A Comprehensive Database of Protein Dynamics. Structure 18, 423–435 (2010).
Grüning, B. et al. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods 15, 475–476 (2018).
da Veiga Leprevost, F. et al. BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics (Oxford, England) 33, 2580–2582 (2017).
Ison, J. et al. EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics (Oxford, England) 29, 1325–1332 (2013).
Hillion, K.-H. et al. Using bio.tools to generate and annotate workbench tool descriptions. F1000Research 6, ELIXIR-2074 (2017).
Doppelt-Azeroual, O. et al. ReGaTE: Registration of Galaxy Tools in Elixir. GigaScience 6, 1–4 (2017).
Carter, D., He, J., Rubble, J. R. & Wright, B. The structure of the orthorhombic form of hen egg-white lysosome at 1.5 angstroms resolution. Protein Data Bank, Rutgers University, https://identifiers.org/pdb:1AKI (1997).
Valentini, G. et al. Human erythrocyte pyruvate kinase. Protein Data Bank, Rutgers University, https://identifiers.org/pdb:2VGB (2007).
Canu, G., De Bonis, M., Minucci, A. & Capoluongo, E. Red blood cell PK deficiency: An update of PK-LR gene mutation database. Blood Cells, Molecules, and Diseases 57, 100–109 (2016).
wwPDB consortium. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Research 47, D520–D528 (2018).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Hornak, V. et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 (2006).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry 18, 1463–1472 (1997).
The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research 47, D506–D515 (2018).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935 (1983).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N · log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 52, 7182–7190 (1981).
Andrio, P. et al. Bioexcel building blocks test cases. Zenodo. https://doi.org/10.5281/zenodo.2581362 (2019).
Acknowledgements
We thank The Barcelona Supercomputing Center for providing all the HPC resources to launch the test executions presented in this manuscript. The project was supported by the following grants: BioExcel Center of Excellence (Horizon 2020 Framework Programme. Grants 675728, and 823830). Elixir-Excelerate (Horizon 2020 Framework Programme. Grant 676559). Spanish National Institute for Bioinformatics (Institute of Health Carlos III. Grants PT13/0001/0019, PT13/0001/0028, PT17/0009/0001). Spanish Government, Severo Ochoa Grant SEV2015-0493, TIN2015-65316-P, Generalitat de Catalunya. 2014-SGR-1051, 2017-SGR-1110.
Author information
Authors and Affiliations
Contributions
J.L.G. designed the strategy and wrote the manuscript with contributions of all authors. P.A. and A.H. were responsible of software development, J.C., D.L. and R.M.B. were responsible of PyCOMPSs adaptation and implementation, L.J. developed and validated HPC workflows, M.P. and L.C. developed and tested the Galaxy implementation. S.S.-R. helped to implement CWL and Jupyter Notebook. C.G., R.M.B., M.O. check the manuscript and provided useful additions.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andrio, P., Hospital, A., Conejero, J. et al. BioExcel Building Blocks, a software library for interoperable biomolecular simulation workflows. Sci Data 6, 169 (2019). https://doi.org/10.1038/s41597-019-0177-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0177-4
This article is cited by
-
RETRACTED ARTICLE: Application of intelligent measuring robot in engineering cost management
International Journal of System Assurance Engineering and Management (2023)
-
pyKVFinder: an efficient and integrable Python package for biomolecular cavity detection and characterization in data science
BMC Bioinformatics (2021)
