
Abstract
A new methodology termed selective organ targeting (SORT) was recently developed that enables controllable delivery of nucleic acids to target tissues. SORT lipid nanoparticles (LNPs) involve the inclusion of SORT molecules that accurately tune delivery to the liver, lungs and spleen of mice after intravenous administration. Nanoparticles can be engineered to target specific cells and organs in the body by passive, active and endogenous targeting mechanisms that require distinct design criteria. SORT LNPs are modular and can be prepared using scalable, synthetic chemistry and established engineering formulation methods. This protocol provides detailed procedures, including the synthesis of a representative ionizable cationic lipid, preparation of multiple classes of SORT LNPs by pipette, vortex and microfluidic mixing methods, physical characterization, and in vitro/in vivo mRNA delivery evaluation. Depending on the scale of the experiments, the synthesis of the ionizable lipid requires 4–6 d; LNPs can be formulated within several hours; LNP characterization can be completed in 2–4 h; and in vitro/in vivo evaluation studies require 1–14 d, depending on the design and application. Our strategy offers a versatile and practical method for rationally designing nanoparticles that accurately target specific organs. The SORT LNPs generated as described in this protocol can therefore be applied to multiple classes of LNP systems for therapeutic nucleic acid delivery and facilitate the development of protein replacement and genetic medicines in target tissues. This protocol does not require specific expertise, is modular to various lipids within defined physicochemical classes, and should be accomplishable by researchers from various backgrounds.
Similar content being viewed by others
Introduction
Development of the protocol
The vast potential and practical application of messenger RNA (mRNA) technology was realized during the past 2 years through the development of multiple coronavirus SARS-CoV-2 mRNA lipid nanoparticle (LNP) vaccines for COVID-19. Compared with other functional biomolecules, mRNA offers unique speed, modularity and safety due to the ability to rapidly design mRNA sequences, package mRNAs into tunable delivery systems, and mediate transient activity in a dose-controllable manner. These benefits make mRNA highly exciting for the development of a new generation of medicines1,2. In 2020, Pfizer–BioNTech and Moderna developed safe and effective COVID-19 mRNA LNP vaccines that have saved lives3,4. The notable use of LNP technology to deliver the mRNA-encoded antigen into cells stimulates discussion of what other therapies may be deployed by LNP technologies. Standing out across the pharmaceutical industry as a clinically mature and effective technology to deliver nucleic acids2,5,6,7,8,9,10, LNPs have emerged as the most promising delivery approach for new vaccines11,12,13,14,15,16, protein replacement therapies17,18,19,20,21, genome editing22,23,24,25,26, cancer immunotherapies27,28,29,30,31,32 and more. Traditional LNPs include four molecules2: an ionizable amino lipid to bind nucleic acids and help with endosomal escape, an amphipathic phospholipid to promote fusion with cell and endosomal membranes33, cholesterol to facilitate LNP stability34 and a polyethylene glycol (PEG) lipid to improve colloidal stability and reduce reticuloendothelial clearance35. Although traditional LNPs are safe and effective, they have been limited to intramuscular administration (as in the case of COVID-19 mRNA LNP vaccines) and intravenous (IV) administration targeting liver hepatocytes (as in the case of Onpattro short interfering RNA LNPs36). The limitation of traditional LNPs largely stems from their physiochemical similarity to very-low-density lipoprotein and propensity to adsorb apolipoprotein E in blood plasma, prompting accumulation in the liver and uptake into hepatocytes via the low-density lipoprotein receptor. These hallmarks, while valuable for liver hepatocyte applications, severely limit applications of traditional LNP technologies outside the liver. The predictable and rational design of LNPs for delivery of nucleic acids to extrahepatic tissues therefore remains a key challenge that must be overcome to reach the full potential of mRNA LNP technologies to allow broad therapeutic development across diverse settings8.
To address this challenge, we developed a methodology termed selective organ targeting (SORT) wherein nanoparticles are systematically engineered to accurately deliver therapeutic molecules to target both hepatic and extrahepatic tissues22. Initial SORT LNPs have been developed to deliver mRNA22,37,38, proteins26 and mixed nucleic acid payloads22,24,26,39 for genome editing to the liver, lungs and spleen following systemic IV administration. SORT LNPs were formed by inclusion of a supplemental SORT molecule22 (Fig. 1a) that facilitates LNP redirection and cellular uptake in target organs via an endogenous targeting mechanism of action40. These effects are dependent on the chemistry and amount of included SORT molecule in a modular and tunable fashion. Our initial development work focused on augmenting basic four-component LNP systems consisting of a degradable dendrimer-based ionizable cationic lipid (5A2-SC8) that was selected from a screen of >1,500 molecules41, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol and 1,2-dimyristoyl-rac-glycero-3-methoxy(poly(ethylene glycol)) (DMG-PEG) and mRNA with a fifth SORT molecule to enable protein replacement and gene correction therapeutics in specific target tissues and cells22. In this approach, addition of a permanently cationic lipid resulted in protein expression predominantly in the lungs, addition of a permanently anionic lipid facilitated delivery to the spleen and addition of an ionizable amino lipid increased delivery to the liver. These SORT LNPs maintain physiochemical stability and consistent in vivo delivery efficacy after storage, are well tolerated and mediate therapeutically relevant levels of protein production22.

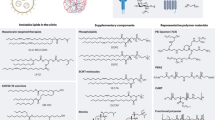
a, Inclusion of a SORT molecule in traditional four-component LNPs (which consist of ionizable cationic lipids, amphipathic phospholipids, cholesterol and PEG lipids) systematically alters the in vivo delivery profile of the resulting five-component SORT LNPs to enable tissue-specific delivery of mRNA to the liver, lungs and spleen of mice after IV administration. b, Chemical structures of the lipids used in this protocol, including the ionizable cationic lipids 4A3-SC8 and DLin-MC3-DMA (MC3), the phospholipids DOPE and DSPC, cholesterol, DMG-PEG and the SORT molecules DOTAP, 18PA and DODAP. c–e, Three mixing methods are described to prepare SORT LNP formulations introduced in this protocol: pipette mixing (c), vortex mixing (d) and microfluidic mixing methods (e). a adapted from ref. 22, Springer Nature Limited. Images in c–e created with BioRender.com.
In this protocol, we focus on examples of two classes of SORT LNPs, aimed for researchers with capacity to conduct chemical synthesis of ionizable amino lipids as well as for researchers who would prefer to conduct studies using commercially available molecules. We first provide instructions to prepare liver-, lung- and spleen-targeting mRNA LNPs that utilize a representative degradable ionizable cationic lipid named 4A3-SC8 synthesized via dendrimer growth orthogonal reactions, which was selected owing to its utility in delivering diverse cargoes in vivo, including siRNA, mRNA, sgRNA and ssDNA38,39,41. Second, we provide instructions to prepare liver-, lung- and spleen-targeting mRNA LNPs that utilize another representative ionizable cationic lipid named DLin-MC3-DMA (MC3), which was selected owing to its use in Onpattro36 and commercial availability. 4A3-SC8 can be formulated with other lipids to form an mRNA-loaded dendrimer lipid nanoparticle (mDLNP) system with validated efficacy in traditional liver delivery applications. The base four-component mDLNP formulation herein consists of 4A3-SC8, DOPE, cholesterol and DMG-PEG at the molar ratio of 23.8/23.8/47.6/4.8, and mRNA (total lipids/mRNA = 40/1, wt/wt). To form SORT LNPs, SORT molecules are added to the above-established LNP formulation in specific amounts, while keeping the relative molar ratio among those four components constant to maintain effective RNA encapsulation and endosomal escape capabilities. According to previous experience, the tuning of tissue tropism may be dependent on specific chemical functional groups and the physicochemical (e.g., permanently cationic, anionic, ionizable cationic, zwitterionic, etc.) properties of the SORT molecule and the amount of added SORT molecule, leading to predictable and controllable mRNA translation to protein in one organ or in a combination of multiple organs22. Thus, it is important to keep in mind that both the chemistry and the amount of added SORT molecule will affect delivery outcomes. Nevertheless, selection of lipids is a modular process, wherein similar delivery outcomes are expected when substituting molecules defined within the general classes of permanently cationic, anionic, ionizable cationic, and zwitterionic lipids. For the purposes of this protocol, we firstly prepared liver-targeting 4A3-SC8-based four-component mDLNPs, then added SORT molecules to create liver-targeting five-component 4A3-SC8-based SORT LNPs that include an tertiary amino lipid 1,2-dioleoyl-3-dimethylammonium-propane (DODAP) at 20% molar ratio of the total lipids (that was found to enhance delivery specificity and potency in the liver22), lung-targeting 4A3-SC8-based SORT LNPs that include a quaternary amino lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) at 50% molar ratio of the total lipids, and spleen-targeting 4A3-SC8-based SORT LNPs that include 1,2-dioleoyl-sn-glycero-3-phosphate (18PA) at 10% molar ratio of the total lipids. To demonstrate this approach is applicable to other types of nanoparticle systems, we also formulated MC3 with 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, DMG-PEG at a molar ratio of 50/10/38.5/1.5, and mRNA (total lipids/mRNA = 40/1, wt/wt) to obtain liver-targeting base four-component MC3 LNPs. MC3-based SORT LNPs were generated with the addition of either 50 mol% (e.g., 50% molar ratio of the total lipids) of DOTAP to target the lungs or 30 mol% of 18PA to target the spleen. For chemical structures of all the lipids used in this protocol, see Fig. 1b.
As for making SORT LNP formulations, formation of LNPs occurs via the ethanol dilution method wherein one equivalent volume of the ethanol solution containing all the lipids is rapidly mixed with three equivalent volumes of the aqueous buffer solution containing mRNA. There are several employed methods to realize rapid mixing of these two phases (Fig. 1c): the pipette mixing method, the vortex mixing method and the microfluidic mixing method. Pipette mixing is the most common method to prepare small-scale batches of LNPs for optimization, characterization, in vitro studies and low-dose in vivo studies, which involves simply mixing two solutions by pipetting up and down by hand rapidly for a specific time to allow the formation of nanoparticles. This method is easy to operate, low cost and generally appropriate for scales from ~6/18 to ~30/90 (µL/µL, ethanol/aqueous). Vortex mixing is a second method to prepare medium-scale batches of LNPs for optimization, characterization, in vitro studies and in vivo studies, which involves rapid mixing of the two solutions under vigorous vortexing for a specific time to allow the formation of nanoparticles. This method is also easy to implement and execute at a laboratory scale but does require a vortex mixer, which is relatively inexpensive. It is generally appropriate for medium scales from ~20/60 to ~40/120 (µL/µL, ethanol/aqueous). These first two methods are widely accessible and can be carried out under ordinary laboratory conditions. Microfluidic mixing42,43,44,45 is a third method to prepare medium- to large-scale batches of LNPs for in vitro studies, in vivo studies and clinical use. Laboratories commonly use homemade custom microfluidic mixing devices that utilize microfluidic mixing channels (often made via polydimethylsiloxane microfluidic fabrication) connected to commercially available syringe pumps. Laboratories can also use commercial mixing systems, such as the NanoAssemblr from Precision Nanosystems. To manufacture consistent batches for in vivo animal studies or potential clinical use where large scales are required, the microfluidic mixing method offers notable advantages including reproducibility, LNP uniformity and controllability of mixing parameters that can modulate LNP properties such as size and consistency over a broad scale range. In this protocol, we employed the NanoAssemblr technology wherein lipids in ethanol and mRNA in aqueous solution were pumped into the two inlets of the cartridge. The chaotic advection of the laminar streams induced by the herringbone structures in the cartridge caused rapid mixing of the two solutions and yielded highly reproducible nanoparticles with limited sizes44. This method is more technical in nature and expensive, requiring researchers to either build a microfluidic device or purchase a microfluidic mixing instrument. It is generally appropriate for medium-to-large scales from 1–20 mL (e.g., NanoAssemblr Ignite) to 0.01–1 L (e.g., NanoAssemblr Blaze). All three methods follow the general principle of rapid mixing: LNPs are formed by a quick increase in polarity of the medium induced by rapid mixing of two miscible phases in a controlled environment. This rapid mixing induces supersaturation of lipid molecules, which leads to the self-assembly of LNPs. However, they possess respective advantages and drawbacks. It is important to select the appropriate method to prepare LNPs for mRNA delivery to specific tissues according to scale requirements, tolerance of batch-to-batch variation, suitability of operations, availability of specialized equipment and ultimate end application needs.
Herein, we provide protocols to prepare 4A3-SC8- and MC3-based liver, lung and spleen SORT LNPs via three methods (pipette mixing, vortex mixing and microfluidic mixing) to provide readers with options to cover small-, medium- and large-scale production. Each formulation is then characterized and evaluated for in vivo delivery of mRNA to provide references for expected results. We anticipate that these general methods will be easily adaptable for researchers using other lipids within the defined physiochemical classes.
Applications of the method
Despite substantial research and development of LNPs, detailed guides for the preparation of mRNA LNPs have not been published to our knowledge. Here we focus on 4A3-SC8- and MC3-based SORT LNP formulations owing to their established in vivo efficacy and broad utility to deliver diverse cargoes, further utilizing DOTAP, 18PA and DODAP as representative SORT molecules. We note that the SORT strategy is applicable to multiple classes of LNP systems—SORT LNPs are modular with respect to each of the ‘classes’ or categories of molecules of which they are composed; thus, one can substitute one ionizable amino lipid for another, or one phospholipid for another, for example, and achieve comparable results; moreover, many eligible SORT molecules with different percentages can be chosen for diverse purposes22. Other permanently cationic, anionic or ionizable amino lipids can alternatively be used as lung, spleen and liver SORT molecules, respectively22. SORT is further compatible with, and able to, deliver various therapeutics, including nucleic acids, proteins and genome editors. As diseases causing DNA mutations occur in specific cells and organs, one unique advantage of SORT LNPs is their ability to enable tissue-specific genome editing. For example, liver, lung and spleen SORT LNPs were used to accurately deliver Cas9 mRNA/single guide RNA or Cas9 ribonucleoprotein complexes for gene editing in the liver, lungs, spleen, muscle and brain8,22,26. Therapeutic applications included gene knockouts in the liver for cardiovascular disease treatment and restoration of dystrophin expression for Duchene muscular dystrophy22,26. Liver and lung SORT LNPs were also used to create genetically defined organ-specific tumor models via in situ genome editing26. It is exciting that LNPs can be engineered straightforwardly to enable therapeutic gene regulation in specific tissues and cells, as this approach holds promise to create target medicines for diseases such as cystic fibrosis (lung), cancer via CAR-T therapy (spleen), immune disorders and transthyretin amyloidosis (liver). We also envision this methodology may be further expanded to allow the rational design of nanoparticles that selectively target specific tissues for the delivery of a variety of drug types and additional tissues in the future.
Alternative methods
Chemical, biological and physical methods can be applied to achieve delivery of mRNA to specific tissues to some extent. For example, LNPs can be nebulized to provide local and direct delivery of mRNA to the lungs. In addition to aerosol delivery to the lungs, intrathecal administration may be useful for delivery to cerebrospinal fluid reaching the brain, and intramuscular administration can allow mRNA LNPs to transfect muscle cells locally. These administration methods generally provide a high local concentration of mRNA LNPs and greatly reduce systemic exposure. In contrast, chemical methods are more diverse and may allow mRNA LNPs to reach deep tissues with broader penetration. mRNA LNPs for systemic delivery to tissues and tumors could employ passive, active and endogenous targeting strategies. The mechanism of SORT LNP selectivity is hypothesized to be endogenous targeting40. Alternatively, active targeting LNPs have been developed wherein the surface of LNPs was modified with ligands or cell-specific antibodies that bind to, or are internalized by, specific receptors expressed on the target cells. However, this approach is limited by the lack of tumor-specific surface markers and complexity in chemical synthesis for each LNP. Other efforts have involved using PEGylation to increase blood circulation time, control size and promote LNP accumulation in tumor tissues after systemic delivery via passive targeting46. The unique tumor environment characteristics may also be exploited by pH-responsive nanoparticles to increase tumor delivery47,48.
With respect to SORT LNP formulation, the methods described involving pipette, vortex and microfluidic mixing all require a rapid mixing speed to quickly increase the polarity of the environment, which benefits the formation of uniform LNPs. There are also some additional techniques that can be applied for LNP production. The conventional thin-film hydration method and subsequent postprocessing methods such as extrusion and sonication are common manufacturing methods for preparing lipid complexes and liposomes49,50, and have been widely used in development of drug-loaded liposomes, but have been less commonly applied to LNPs. Lipids are dissolved in an organic solvent in a vessel or a round-bottom flask where the organic solvent is removed in vacuo and a lipid film is left on the surface. Large multilamellar vesicles are typically formed upon hydration with an aqueous solution including nucleic acids, and then are rearranged into small unilamellar vesicles by size-reduction steps such as extrusion or sonication. However, the multiple steps are time consuming; extrusion of large lipid vesicles may clog the membrane, resulting in product losses; and sonication can lead to oxidation and degradation of lipids. Therefore, although this method is still widely used, some disadvantages hinder its development and scale-up.
Finally, we note that commercial LNPs, such as the mRNA LNP COVID-19 vaccines, are manufactured via a fourth option termed T-mixing, in which organic and aqueous phases are mixed in a controlled manner for producing LNPs49. Commercial-grade pumps flow ethanol and aqueous solutions through a T-shaped mixing design, and the collision of the two input streams in the T-junction causes rapid mixing, resulting in a turbulent output flow and quick increase of the solvent polarity. Increasing flow rates lead to smaller particle size, and the encapsulation efficiencies are generally higher compared with conventional methods44,51,52. However, like the microfluidic method, T-junction mixing is not appropriate for small laboratory-scale batches but is preferred for production on a large scale by companies engaged in LNP manufacturing owing to compatibility with continuous flow streams, and scalability due to the use of continuous flow methods (no pressure or volume constraints). It is not conducive to operating at scales suitable for common laboratory experiments such as for in vitro testing; therefore, we have not included it in this protocol.
Expertise needed
This protocol provides a detailed and coordinated process of lipid chemical synthesis, SORT LNP formulation, in vitro studies and in vivo studies, which is streamlined for ease of use and adaptable across three methods for different scales. Researchers with backgrounds in chemistry, biochemistry, molecular biology, pharmaceutical sciences, biomedical sciences and engineering should be able to perform this protocol. Regarding chemical synthesis, an organic chemistry lab can assist in reproducing these compounds if chemical fume hoods and glassware are not accessible in biological labs. For LNP characterization, a biomedical engineering lab can assist with physical methods provided that the required equipment is available. Microfluidic experiments must be performed in a lab equipped with the relevant instruments.
Limitations
SORT methodology
Although the SORT methodology has achieved great success in tissue-specific delivery and has the potential to be applied to diverse material classes, it has so far been utilized only in LNP systems. Our previous study reported that supplementing a completely inactive LNP formulation with DODAP or DOTAP resulted in tissue-specific delivery to the spleen and lungs22. It is noted that all the SORT LNPs contain ionizable cationic lipids, which are considered essential for endosomal escape owing to their ability to acquire charge after endocytosis and are needed for efficacy. Other types of LNP systems or polymeric systems may follow a different mechanism, and more data are needed. It is further noted that the percentage and biophysical properties of the SORT molecule play an important and specific role in altering in vivo delivery profile40. The selection of SORT molecules covers a wide range of known molecules, and an examination of an expanded set of eligible chemicals is currently under study. SORT LNPs are currently optimized for five-component LNP systems; however, four-component SORT LNPs have been successfully prepared38, and the SORT concept may be applied in the future to additional mixtures of lipids and polymers all while using SORT molecules for endogenous targeting. Moreover, the SORT strategy can currently only realize organ-specific delivery to the liver, spleen and lungs. It is conceivable that SORT may allow delivery to additional tissues, though approaches to deliver RNA-LNP therapeutics to other organs, tumor tissues and specific cells still need to be explored and developed.
Technical methods to prepare SORT LNP formulations
Despite the aforementioned advantages these three methods (pipette, vortex and microfluidic mixing) in the protocol have in preparing SORT LNP formulations, their limitations should also be considered. The pipette and vortex mixing methods are labor-intensive production processes thereby confined to laboratory batch productions that are difficult to scale up. Since they are still macroscopic mixing methods, the resulting particle sizes are relatively larger and the reproducibility between batches is considered improvable. The microfluidic mixing method (e.g., the NanoAssemblr technology used in this protocol) is preferred for large-scale production for laboratory animal studies (all areas of research, including academia and industry), in companies and for preclinical and clinical uses. However, microfluidic mixing is difficult to reconcile with very small laboratory-scale batches. There is a dead volume in preparation, which causes waste of lipid materials and nucleic acids, making the approach cost prohibitive for some cases and studies. In addition, overusing a cartridge may jeopardize experimental results owing to clogging that causes material loss, cross-contamination and instrument damage, making it more costly to maintain the instruments and purchase the equipment parts. Microfluidic mixing yielded the highest consistency, smallest LNP sizes and highest efficacy (‘Anticipated results’). Overall, we generally recommend hand pipette mixing for high-throughput in vitro studies, hand pipette and vortex mixing for general in vitro studies, and vortex mixing and microfluidic mixing for in vivo studies. Having said that, all three methods can be used for all applications.
Experimental design
Overview
In this protocol, we present step-by-step procedures for (i) synthesis of the 4A3-SC8 ionizable lipid (Steps 1–25), which is an efficacious ionizable cationic lipid selected from our library41; (ii) preparation of selective organ-targeting (SORT) LNPs using 4A3-SC8 or MC3 as the base cationic ionizable lipid for tissue-specific mRNA delivery by three technical methods: pipette, vortex and microfluidic mixing methods (Step 26 and Boxes 1 and 2) and characterization (Step 27); (iii) in vitro Luc mRNA delivery (Steps 28–36); and (iv) systematic comparison of the in vivo Luc mRNA delivery and expression efficacies by SORT LNPs prepared by three different methods (Steps 37–41). Overall, this protocol will provide researchers with detailed and practical instructions to prepare LNPs for lung-, spleen- and liver-targeted mRNA delivery (see flow chart in Fig. 2).

Steps 1–25 outline synthesis and purchase of lipids; Step 26 outlines preparation of SORT LNP formulations; Step 27 outlines characterization; Steps 28–41 outline in vitro/in vivo delivery of Luc-mRNA by SORT LNPs.
Synthesis of the ionizable amino lipid 4A3-SC8
The development of effective and nontoxic delivery systems remains an important goal for enabling broad clinical application of mRNA therapeutics. Eligible and efficacious delivery systems should include the following essential elements: chemical groups to efficiently bind mRNA during mixing and release mRNA after endocytosis, nanoparticle-stabilizing hydrophobicity, and low toxicity. Chemical features to achieve these goals frequently include tertiary amines with optimal pKa (~6.4), lipid-like hydrophobic domains (e.g., alkyl and alkenyl chains), well-defined monodisperse structures and degradable bonds (e.g., esters) that collectively result in safe degradation into metabolizable or excretable fragments. These factors allow LNPs to overcome a series of extracellular and intracellular barriers in a safe and effective manner. To achieve the balance of high potency and low toxicity, we previously designed and screened a library of ionizable cationic lipids using orthogonal dendrimer growth chemistry that contained a variety of chemically diverse RNA-binding amino cores and alkyl branches linked by biocompatible and degradable esters41. Further studies demonstrated that low-generation G1 dendrimers with a binding core and four to six branches with SC8–12 periphery composition exhibited the highest potency for in vivo RNA delivery when formulated with helper lipids, cholesterol and PEG lipids. Numerous molecules from this library including 5A2-SC8 that was used for initial development of SORT, and many variations based on cores such as but not limited to 2A2, 3A3, 3A4, 4A1, 4A3, 5A1, 5A2, 5A3, 5A4, 6A1 and 6A3 with various hydrophobic domains (such as SC5 to SC14), have found utility across various applications22,26,37,38,39,41,53.
In this protocol, we use 4A3-SC8 (2) as an outstanding representative lipid (Fig. 3) owing to the relative ease of synthesis, high efficacy profile and adaptability to deliver diverse cargoes38,39,41. The synthesis of 4A3-SC8 conjugates the 4A3 amino core with the SC8 alkyl branches through sequential aza- and sulfa-Michael additions by an asymmetric and degradable monomer 2-(acryloyloxy)ethyl methacrylate (AEMA, 1). AEMA is designed to have an acrylate group on one end and a methacrylate group on the other to selectively react with amine and thiol functionalities, respectively. To be more specific, the 4A3 amine core with four N–H bonds first reacted quantitatively with the less steric acrylate group of AEMA at 50 °C for 24 h in the presence of 5 mol% of butylated hydroxytoluene (BHT) to avoid radical polymerization. The product then reacted with 1-octanethiol (SC8) under optimized dimethylphenylphosphine (DMPP)-catalyzed conditions at 55 °C for 48 h. The resulting product was purified by flash chromatography to obtain the final product 4A3-SC8 (2). This methodology is consecutive and easy to operate. We provide a detailed protocol in the ‘Procedure’ section for the synthesis of AEMA (Steps 1–14) and 4A3-SC8 (Steps 15–25). One can also fine-tune the physicochemical properties of the delivery carriers by modulating the topological structures of the ionizable amine cores or the alkyl branches41.

TEA: triethylamine; CHCl3: chloroform; BHT: butylated hydroxytoluene; DMPP: dimethylphenylphosphine; RT: room temperature.
Preparation of SORT LNPs by three different technical methods
The base 4A3-SC8 mDLNP formulation consisting of 4A3-SC8, DOPE, cholesterol and DMG-PEG at the molar ratio of 23.8/23.8/47.6/4.8 with mRNA (total lipids/mRNA = 40/1, wt/wt) was supplemented with a SORT molecule without changing the original relative molar ratios of the basic four components. On the basis of the four-component 4A3-SC8 mDLNP formulation, the addition of 20 mol% DODAP, 50 mol% DOTAP or 10 mol% 18PA, respectively, resulted in five-component liver, lung or spleen SORT LNPs. To demonstrate that this approach is applicable to other types of nanoparticle systems, we also formulated the MC3 ionizable amino lipid with DSPC, cholesterol and DMG-PEG at the molar ratio of 50/10/38.5/1.5 with mRNA (total lipids/mRNA = 40/1, wt/wt) to obtain base MC3 LNPs. MC3 was selected owing to its use in Onpattro32 and commercial availability. MC3-based SORT LNPs were then generated with the addition of either 50 mol% of DOTAP or 30 mol% of 18PA.
Before formulating self-assembled SORT LNPs, all lipids with specified molar ratios were dissolved and mixed in ethanol to form a complete lipid mix solution. Separately, mRNA, e.g., luciferase-encoding mRNA (Luc mRNA, used as a reporter gene mRNA), was dissolved in 10 mM citrate buffer (pH 4). In the specific case of spleen SORT LNP formulations, 18PA was first dissolved in tetrahydrofuran and ethanol (1/1, vol/vol), then mixed with all other lipid components dissolved in ethanol. Separately for spleen SORT LNPs, Luc mRNA was dissolved in 10 mM citrate buffer (pH 3). Then, for all standard and all SORT LNPs, the ethanol solution containing all lipids was rapidly mixed with the aqueous buffer solution containing Luc mRNA at a ratio of 3/1 (aqueous/ethanol, vol/vol) to achieve a final weight ratio of 40/1 (total lipids/mRNA, wt/wt). We note that lower ratios (e.g., weight ratio of 20/1 (total lipids/mRNA, wt/wt)) are also effective, which can be valuable when higher mRNA doses are needed for specific studies to reduce potential lipid toxicity. We provide details in the ‘Procedure’ section (Step 26). In this protocol, we also performed three common technical methods (pipette, vortex and microfluidic mixing methods) to make SORT LNP formulations. One can make appropriate selection depending on reasonable requirements and applications, varying from bench-scale batches to larger-scale batches for animal studies. Instructions for applying these three methods are in the ‘Procedure’ section.
Characterization of SORT LNPs
After SORT LNPs are formed, the fresh LNPs were diluted tenfold (1 ng/µL mRNA concentration) in RNase-free water respectively for particle size measurements on a Malvern Zetasizer Nano ZS machine. The encapsulation efficiency of mRNA in each LNP was quantified by measuring the mRNA binding following the Quant-iT RiboGreen assay protocols.
In vitro delivery of Luc mRNA
Two base mRNA delivery LNP formulations (4A3-SC8 mDLNPs and MC3 LNPs) prepared by the pipette mixing method were used as examples to provide a protocol for in vitro experiments. Luc mRNA LNP formulations were diluted to 1 ng/µL mRNA concentration and used to evaluate the delivery to ovarian cancer cells IGROV-1, which is a representative cell line with moderate resistance to transfection. A proprietary RNA interference (RNAi)-specific cationic lipid formulation Lipofectamine RNAiMAX was used as a positive control. The preparation details are in the ‘Procedure’ section (Step 31). IGROV-1 cells were treated with 4A3-SC8 mDLNPs, MC3 LNPs and RNAiMAX formulations containing Luc mRNA (25 ng mRNA per well, n = 5 per group) for 24 h before cell viability and luciferase gene expression were measured using the ONE-Glo + Tox luciferase reporter and cell viability assay.
In vivo delivery of Luc mRNA by SORT LNPs prepared by three methods
C57BL/6 mice (female, 6–8 weeks old, ~20 g) were intravenously injected with various SORT LNP formulations prepared by pipette, vortex, or microfluidic mixing methods at an mRNA dose of 0.1 mg/kg (n = 3 per group) to evaluate and compare the in vivo Luc mRNA delivery and luciferase protein expression efficiencies. After 6 h, mice were injected intraperitoneally with D-luciferin solution and the ex vivo images of luminescence in major organs were obtained and quantified using an IVIS Lumina system.
Materials
Biological materials
Caution
All the animal experiments were approved by the Institution Animal Care and Use Committee (IACUC) of the University of Texas Southwestern Medical Center (UTSW) and were consistent with local, state and federal regulations as applicable. Mice were housed in a barrier facility with a 12 h light/dark cycle and maintained on standard chow (2916 Teklad Global). The temperature range for the housing room is 68–79 °F (average is ~72 °F), and the humidity range is 30–50% (average is ~50%).
-
C57BL/6 mice (female, 6–8 weeks old, purchased from Charles River Laboratories and maintained in the UTSW Animal Facility)
Critical
For the ease of comparison, all the mice used in this protocol were female mice of the same age. However, our previous studies indicate that the sex of animals does not affect the results22. It is worth noting that the weight of adult male mice is on average higher than that of adult female mice of the same age. The injection doses should be adjusted depending on the actual weights of the mice to control accurate mg/kg mRNA dosing.
-
IGROV-1 cells (Sigma-Aldrich, cat. no. SCC203); RRID: CVCL_1304;
Caution
We ensured that all the cells used in this protocol had no issues of cell line misidentification and cross-contamination according to the latest report provided by the International Cell Line Authentication Committee (http://iclac.org/databases/cross-contaminations), and they are free of mycoplasma.
Reagents
Caution
Most of the reagents and solvents used in the protocol require the use of protective goggles, gloves and lab coats
Critical
It is strongly recommended that all the reactions be performed in a fume hood. Solid and liquid waste generated during all the procedures should be disposed of properly, according to institutional and government guidelines.
-
Acryloyl chloride, ≥97% (Sigma-Aldrich, cat. no. A24109)
Caution
Acryloyl chloride is harmful and smells malodorous. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
Chloroform (CHCl3; anhydrous; Fisher Scientific, cat. no. AC610281000)
Caution
Chloroform is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
Cholesterol, ≥99% (Sigma-Aldrich, cat. no. C3045)
-
Citric acid, 99% (Sigma-Aldrich, cat. no. C0759)
-
Deionized water (facilitated by UT Southwestern)
-
Deuterated chloroform (CDCl3, 99.9%, extra dry, stabilized; Fisher Scientific, cat. no. AC610281000)
Caution
Deuterated chloroform is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
2,6-Di-tert-butyl-4-methylphenol (BHT; Sigma-Aldrich, cat. no. B1378)
-
3,3′-Diamino-N-methyldipropylamine (4A3; Sigma-Aldrich, cat. no. B0821)
-
DMPP (dimethylphenylphosphine), 99% (Sigma-Aldrich, cat. no. 265020)
Caution
DMPP is harmful and smells malodorous. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
DLin-MC3-DMA (MC3; MedKoo Biosciences, cat. no. 555308)
-
DMG-PEG (1,2-dimyristoyl-rac-glycero-3-methoxy(poly(ethylene glycol)) (Avanti Polar Lipids, cat. no. 880151)
-
DODAP (1,2-dioleoyl-3-dimethylammonium-propane) (Avanti Polar Lipids, cat. no. 890850)
-
18PA (1,2-dioleoyl-sn-glycero-3-phosphate (sodium salt)) (Avanti Polar Lipids, cat. no. 840875)
-
DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) (Avanti Polar Lipids, cat. no. 850365)
-
DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) (Avanti Polar Lipids, cat. no. 850725)
-
DOTAP (1,2-dioleoyl-3-trimethylammonium-propane (chloride salt)) (Avanti Polar Lipids, cat. no. 890890)
-
Ethanol (EtOH, 200 Proof; Fisher Scientific, cat. no. BP2818500)
Caution
Ethanol is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
Ethyl acetate (EA; Fisher Scientific, cat. no. E195)
Caution
EA is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
FBS (Gibco, cat. no. 26140-079)
-
Hexane (Fisher Scientific, H302)
Caution
Hexane is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
2-Hydroxyethyl methacrylate, 97% (Sigma-Aldrich, SKU: 128635-500G)
-
Isoflurane (RxElite, cat. no. NDC60307-120-25)
Caution
Isoflurane is a profound respiratory depressant. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle isoflurane inside a hood when appropriate.
-
Lipofectamine RNAiMAX (Invitrogen Lipofectamine RNAiMAX Transfection Reagent; Thermo Fisher Scientific, cat. no. 13778075)
-
Luciferase mRNA (Luc mRNA, CleanCap Firefly Luciferase mRNA (5-methoxyuridine), 1.0 mg/mL; Trilink, SKU: L-7202-1000)
-
d-Luciferin, sodium salt (Goldbio, cat. no. LUCNA)
-
SC8 (1-octanethiol), ≥98.5% (Sigma-Aldrich, cat. no. 471836)
Caution
SC8 is harmful and smells malodorous. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
ONE-Glo + Tox Luciferase Reporter and Cell Viability Assay Kit (Promega, cat. no. E7120)
-
PBS (10×), sterile-filtered (Sigma-Aldrich, cat. no. D1408)
-
PBS (1×), sterile-filtered (Sigma-Aldrich, cat. no. D8537)
-
Quant-it RiboGreen RNA Assay Kit (Invitrogen, cat. no. R11490)
-
Sodium bicarbonate (NaHCO3, ≥99.7%; Sigma-Aldirch, cat. no. S6014)
-
Sodium chloride (NaCl, >99.0%; Sigma-Aldrich, cat. no. S9998)
-
Sodium citrate dihydrate (≥99.0%; Sigma-Aldrich, cat. no. W302600)
-
Sodium sulfate (Na2SO4, ≥99.0%; Sigma-Aldrich, cat. no. 238597)
-
RPMI-1640 medium (1×; CORNING, cat. no. 10-041-CV)
-
Triethylamine (TEA, ≥99.5%; Sigma-Aldrich, cat. no. 471283)
Caution
TEA is highly volatile and smells malodorous. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
UltraPure DNAse/RNase-free distilled water (Fisher Scientific, cat. no. 10-977-023)
Equipment
-
Access to an NMR spectrometer Bruker AN400 (for details, see https://www.utsouthwestern.edu/labs/nmr/instruments-rates/).
-
Addition funnels
-
Anesthesia instrument with isoflurane and O2 flow
-
Balloons
-
Bath sonicator
-
Beakers
-
Benchtop microfluidic mixing platform (NanoAssemblr Benchtop, Precision Nanosystems)
Critical
The NanoAssemblr Benchtop has been discontinued. Please see replacement models such as the Ignite at https://www.precisionnanosystems.com.
-
BRAND UV polystyrene disposable cuvette (Sigma-Aldrich, cat. no. Z637092)
-
Buchner filter funnels
-
Cartridges for Nanoassemblr
-
Centrifuge tubes (15 and 50 mL)
-
Conical flasks
-
Dialysis kits (Pur-A-Lyzer Midi 3500 Dialysis Kit; Sigma, SKU no. PURD35100-1KT. Pur-A-Lyzer Maxi 3500 Dialysis Kit; Sigma, SKU no. PURX35015-1KT)
-
Dissection kit
-
Distillation instruments
-
Erlenmeyer flasks
-
Freezer (−80 °C and −20 °C) and refrigerator (4 °C)
-
Glass capillary spotting tubes
-
Glass collecting tubes (VWR Culture Tubes, Disposable, Borosilicate Glass)
-
Glass vials with caps (4, 10 and 20 mL)
-
Ice-water bath
-
37 °C incubator supplemented with 5% CO2 atmosphere
-
Insulin syringes (29 G 1/2)
-
Magnetic stirring bars
-
Magnetic stirring hot plates
-
Microplate reader (TECAN Infinite 200 PRO)
-
1.5 mL tubes (Eppendorf, RNAse/DNAse free)
-
NanoDrop fluorospectrometer (Thermo Scientific NanoDrop 3300 Fluorospectrometer)
-
Needles for single use
-
Neutral alumina column
-
Nitrogen gas
-
NMR tubes
-
Oil bath
-
Perkin Elmer Xenogen Lumina In Vivo Imaging System (IVIS)
-
pH meter (Mettler Toledo, SevenExcellence)
-
RediSep Rf Alumina Neutral column (24 g, 40 g and 80 g)
-
Rotary evaporator
-
Rubber stoppers
-
Separatory funnels
-
Single-neck round-bottom flasks (25, 50, 100 and 250 mL)
-
Syringes (1 mL, 5 mL and 10 mL)
-
Teledyne ISCO CombiFlash Rf 200 system
-
TLC plates, silica (Sigma-Aldrich, cat. no. 1057140001)
-
Vacuum and inert gas manifold
-
Vacuum pump
-
Vortex-Genie 2 vortex mixer (Model: SI-0236 (G560))
-
Water bath with temperature control
-
96-Well plate, black and flat bottom (Corning, cat. no. CLS3916)
-
96-Well plate, white opaque and flat bottom (Corning, cat. no. CLS3917)
Reagent Setup
Critical
Unless otherwise indicated, the buffers below can be prepared in advance and stored at 4 °C for several months.
Citrate buffer (0.1 M, pH 3)
Dissolve 0.212 g of sodium citrate dihydrate and 0.630 g of citric acid in 40 mL of RNase-free water. Adjust the pH to 3.0 by adding citric acid and checking the pH on a pH meter.
Citrate buffer (10 mM, pH 3)
Dilute the 0.1 M pH 3 citrate buffer tenfold with RNase-free water (vol/vol).
Citrate buffer (0.1 M, pH 4)
Dissolve 0.482 g of sodium citrate dihydrate and 0.454 g of citric acid in 40 mL of RNase-free water. Adjust the pH to 4.0 by adding sodium citrate dihydrate and checking the pH on a pH meter.
Citrate buffer (10 mM, pH 4)
Dilute the 0.1 M pH 4 citrate buffer tenfold with RNase-free water (vol/vol).
TE buffer (1×)
Dilute the 20× TE buffer from the Quant-it RiboGreen RNA Assay Kit 20-fold with RNase-free water (vol/vol).
5× CellTiter-Fluor AFC reagent
Transfer 10 µL of GF-AFC substrate in 2 mL of assay buffer in the ONE-Glo + Tox Luciferase Reporter and Cell Viability Assay Kit. Mix by vortexing the content until the substrate is thoroughly dissolved.
Critical
The CellTiter-Fluor AFC Reagent should be used within 24 h if stored at room temperature (RT, 20–22 °C). Unused GF-AFC substrate and assay buffer can be stored at 4 °C for up to 7 d with no appreciable loss of activity.
ONE-Glo Luciferase Assay reagent
Transfer the content of one bottle of ONE-Glo buffer to one bottle of ONE-Glo substrate. Mix by inversion until the substrate is thoroughly dissolved. This may require multiple inversions.
Procedure
Part 1: synthesis of AEMA
Timing 1–2 d
-
1
Add a magnetic stir bar to a 500 mL dried single-neck round-bottom flask, and add 120 mL of anhydrous chloroform. Immerse the flask into an ice-water bath (0 °C).
-
2
Add 2-hydroxyethyl methacrylate (26 g, 0.216 mol, 1.0 equiv.), TEA (22 g, 0.228 mol, 1.05 equiv.) and the radical inhibitor BHT (0.48 g, 2.16 mmol, 0.01 equiv.) into the solvent. Dissolve the reagents with stirring.
-
3
Dissolve acryloyl chloride (18.4 mL, 0.228 mol, 1.05 equiv.) in 40 mL of anhydrous chloroform, and transfer them to a 100 mL addition funnel.
-
4
Set up the addition funnel onto the flask through the neck, and use a rubber stopper to seal the funnel on the top. Insert a needle with a balloon filled with anhydrous nitrogen gas through the rubber septum atop the addition funnel. Make sure to purge the environment.
-
5
Add the solution prepared in Step 3 dropwise through the addition funnel into the solution prepared in Step 2 contained in the round-bottom flask under stirring conditions.
-
6
Upon complete addition of the acryloyl chloride solution to the round-bottom flask, remove the flask from the ice-water bath and allow the reaction mixture to stir at RT for 12 h.
-
7
Remove the addition funnel. Then place the round-bottom flask onto a rotary evaporator and remove the chloroform under low pressure at RT. Control the pressure carefully to avoid ‘bumping’ into the collection trap, leaving the desired product in the round-bottom flask. Discard the removed chloroform in an appropriate liquid hazardous waste container.
Caution
Chloroform is hazardous to health. It is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
8
Add 150 mL of hexane into the flask. A precipitate of NH4Cl will appear. Perform a vacuum filtration via a Buchner funnel apparatus consisting of a 186 mL Buchner funnel, 83-mm-diameter-sized ceramic filter and 70-mm-diameter-sized filter paper to remove the precipitate. Discard the precipitate in an appropriate solid hazardous waste container.
-
9
Transfer the liquid filtrate into a 500 mL separatory funnel. Add 150 mL of saturated NaHCO3 solution, and shake the mixture vigorously to mix well. Be careful to feel for any pressure buildup. Turn the separatory funnel upside down, holding the glass stopper securely in place, then slowly vent the flask carefully in the hood with the rotary stopper facing into the hood. Close the stopper, and place the flask into a circular rung stand and allow it to rest for 15 min for separation of the organic, product containing fraction. Open the stopper to discard the bottom-layer aqueous phase in an appropriate liquid waste container, and keep the organic phase in the separatory funnel. Repeat three times from adding saturated NaHCO3 solution onward. Add 150 mL of brine to the last collected organic fraction, and shake the mixture vigorously to mix well. Be careful to feel for any pressure buildup. Turn the separatory funnel upside down, holding the glass stopper securely in place, then slowly vent the flask carefully in the hood with the rotary stopper facing into the hood. Close the stopper, and place the flask into a circular rung stand and allow it to rest for 15 min for separation of the organic fraction. Open the stopper to discard the bottom-layer aqueous phase in an appropriate liquid waste container, and keep the organic phase in the separatory funnel.
Caution
Be careful to watch for any pressure buildups during washing, hold the glassware tightly and carefully, and wear protective goggles, gloves, lab coat and face mask during experiments. Perform all work in a chemical fume hood behind the protective shield.
-
10
Collect the organic fraction in a 500 mL Erlenmeyer flask. Add a magnetic stir bar and 20 g of anhydrous sodium sulfate (Na2SO4) into the flask. Stir the mixture for 3 h at RT to remove residual water from the organic fraction.
-
11
Filter the salts on a Bucher funnel setup. Discard the salts, and collect the filtered organic fraction. Then condense the organic fraction under low pressure using a rotary evaporator to obtain a raw product. Control the pressure carefully to avoid ‘bumping’ into the collection trap, leaving the desired product in the round-bottom flask. Discard the removed hexane in an appropriate liquid hazardous waste container.
Caution
Hexane is hazardous to health. It is highly volatile and flammable. Wear protective goggles, gloves, lab coat and face mask during experiments. Handle it in a fume hood, and dispose it appropriately after use.
-
12
Add ~400 mg of BHT to the raw material. Perform a vacuum distillation to obtain a colorless oil (AEMA, 33.8 g, yield 85%) at 100–120 °C.
Critical step
The distillation temperature can vary depending on the strength of vacuum.
-
13
Add 1% mol of radical inhibitor BHT (~400 mg) into AEMA.
Pause point
AEMA can be stored at −20 °C for several months. Store the compound under nitrogen to avoid moisture that could hydrolyze the ester bonds.
-
14
To characterize AEMA (containing BHT) using 1H NMR, weigh 5 mg of AEMA (containing BHT) in a 1.5 mL tube. Add 500 µL of CDCl3, and vortex it well. Transfer the solution into an NMR tube. Obtain the NMR spectrum on an NMR spectrometer (Bruker AN400). Use software MestReNova to perform the NMR analysis. The chemical structure of AEMA is confirmed by the chemical shift and integration of the peaks.
Part 2: synthesis of 4A3-SC8
Timing 3–4 d
-
15
Add a magnetic stir bar into a 25 mL dried single-neck round-bottom flask.
-
16
Add tetraethylenepentamine (4A3, 1.0 g, 6.885 mmol, 1.0 equiv.), BHT (273 mg, 1.239 mmol, 0.18 equiv.) and then AEMA (5.33 g, 28.94 mmol, 4.2 equiv.) into the flask. Mix them with stirring.
Critical step
The order of addition is important.
-
17
Seal the flask with a rubber stopper. Insert a needle with a balloon filled with anhydrous nitrogen gas through the rubber septum atop the flask. Make sure to purge the environment.
-
18
Immerse the flask into an oil bath at 50 °C, and stir the reaction mixture for 24 h.
-
19
Add DMPP (285 mg, 2.065 mmol, 0.30 equiv.) and SC8 (6.04 g, 41.31 mmol, 6.0 equiv.) to the above reaction mixture prepared in Step 17.
Caution
DMPP and SC8 are harmful and smell malodorous. Handle these chemicals inside a chemical fume hood behind the protective shield, and wear protective goggles, gloves, lab coat and face mask during experiments.
-
20
Repeat Step 17.
-
21
Immerse the flask into an oil bath at 55 °C, and stir the reaction mixture for 48 h.
-
22
Load 1 g of above reaction mixture prepared in Step 20 onto a RediSep Rf 24 g Alumina Neutral column.
Pause point
The leftover reaction mixture can be stored at −20 °C for several months and purified when needed. Alternatively, larger aliquots can be purified with larger capacity columns. We describe here a 1 g purification, which is easy to control and sufficient for the studies described herein. Store the compound under nitrogen to avoid moisture that could hydrolyze the ester bonds.
-
23
Set up the CombiFlash Rf 200 system. Load the crude product on a neutral alumina column. Elute the column with pure hexane first. Next, elute the column with a mixture of EA and hexane. Gradually increase the volume percentage of EA in the mixture from 0% to 5% to remove all the unreacted starting materials and impurities with low polarity. Then increase the volume percentage of EA to 20% to elute the product. Check the eluent in each collecting tube by TLC plates (silica, pure EA, Rf ~ 0.2) when the percentage of EA is 20%. Collect all the pure eluents.
-
24
Remove hexane and EA from the collected eluents under low pressure using a rotary evaporator to provide a colorless and sticky oil (4A3-SC8, 350 mg, yield 44.8%).
Pause point
4A3-SC8 can be stored at −20 °C for several months. Store the compound under nitrogen to avoid moisture that could hydrolyze the ester bonds.
-
25
To characterize 4A3-SC8 using 1H NMR, weigh 5 mg of 4A3-SC8 in a 1.5 mL tube. Add 500 µL of CDCl3, and vortex it well. Transfer the solution into an NMR tube. Obtain the NMR spectrum on an NMR spectrometer (Bruker AN400). Use software MestReNova to perform the NMR analysis. The chemical structure of 4A3-SC8 is confirmed by the chemical shift and integration of the peaks.
Part 3: preparation of selective organ-targeting (SORT) LNPs for tissue-specific mRNA delivery by three methods
-
26
We describe here three general methods to prepare mRNA LNPs. To prepare a small-scale formulation, use the pipette mixing and perform the steps in option A. To prepare a medium-scale formulation, use the vortex mixing method and perform the steps in option B. To prepare a large-scale formulation, use the microfluidic mixing method and perform the steps in option C. The preparation of 4A3-SC8 mDLNP formulation is selected as a typical example, which consists of 4A3-SC8, DOPE, cholesterol, DMG-PEG (23.8/23.8/47.6/4.8) and mRNA (total lipids/mRNA = 40/1, wt/wt). The details of all other formulations are summarized in Tables 1–6.
-
(A)
Preparation of the base 4A3-SC8 mDLNP formulation by the pipette mixing method
Timing 30 min for mixing plus at least 1 h for dialysis
-
(i)
Add 19.44 µL of the 4A3-SC8 mDLNP complete lipid mix solution (prepared in Box 1, step 8) into an RNase-free 1.5 mL tube.
-
(ii)
Add 0.56 µL ethanol into the above tube. Mix well.
-
(iii)
Take another RNase-free 1.5 mL Eppendorf tube, add 52 µL of citrate buffer (10 mM, pH 4) and then add 8 µL of Luc mRNA (1.0 mg/mL) into this second tube. Mix well.
Pause point
The mRNA concentration can be measured by NanoDrop.
-
(iv)
Pipette 60 µL of the mRNA buffer solution from Step 26A(iii) and quickly add it into the lipid mix ethanol solution from Step 26A(ii), followed immediately by pipetting up and down rapidly for 20–30 s (hand mixing).
Critical step
The volume ratio between the aqueous phase and ethanol phase is set to be 3:1. Pipette up and down immediately after mixing the two solutions to prepare uniform LNPs. Deviation from these instructions or slow mixing can yield poorly formed LNPs with reduced activity.
-
(v)
Incubate the resulting solution at RT for up to 15 min.
Pause point
This incubation step is optional. It is recommended to perform the dialysis within 15 min after mixing.
-
(vi)
Dialyze the above solution using a Pur-A-Lyzer Midi 3500 Dialysis tube against 1× PBS for at least 1 h to remove the ethanol and acidic buffers.
-
(vii)
After dialysis, transfer the solution to an RNase-free 1.5 mL tube and measure the volume.
-
(viii)
Compensate the solution with 1× PBS to reach a final volume of 800 µL.
Pause point
The resulting solution can be stored at 4 °C for a few days before use. However, it is recommended to use the formulated LNPs as soon as possible to maintain consistent results. Storage at RT is not recommended. Storage at freezing temperatures is also not recommended unless optimized cryoprotectants are used.
Table 1 4A3-SC8 formulation details for the pipette mixing method Table 2 MC3 formulation details for the pipette mixing method Table 3 4A3-SC8 formulation details for the vortex mixing method Table 4 MC3 formulation details for the vortex mixing method Table 5 4A3-SC8 formulation details for the microfluidic mixing method Table 6 MC3 formulation details for the microfluidic mixing method
-
(i)
-
(B)
Preparation of the base 4A3-SC8 mDLNP formulation by the vortex mixing method
Timing 30 min plus at least 1 h for dialysis
-
(i)
Add 24.30 µL of the 4A3-SC8 mDLNP complete lipid mix solution (prepared in Box 1, step 8) into an RNase-free 1.5 mL tube.
-
(ii)
Add 5.70 µL ethanol into the above tube.
-
(iii)
Take another RNase-free 1.5 mL tube, and add 80 µL of citrate buffer (10 mM, pH 4), then 10 µL of Luc mRNA (1.0 mg/mL). Mix well.
Pause point
The mRNA concentration can be measured by NanoDrop.
-
(iv)
Set the vortex mixer equipment to ‘ON’ status and speed level to ‘1’.
-
(v)
Vortex the mRNA buffer solution from Step 26B(iii) at a moderate speed on the vortex mixer. Then pipette 30 µL of the lipid ethanol mix solution from Step 26B(ii) into the pipette. Quickly add it into the vortexing solution. Continue vortexing the resulting dispersion for another 20–30 s.
Critical step
The volume ratio between the aqueous phase and ethanol phase is set to be 3:1. Quickly add the ethanol phase into the vortexing solution as a single action.
-
(vi)
Incubate the resulting solution at RT for up to 15 min.
Pause point
This incubation step is optional. It is recommended to perform the dialysis within 15 min after mixing.
-
(vii)
Dialyze the above solution using a Pur-A-Lyzer Midi 3500 Dialysis tube against 1× PBS for at least 1 h to remove the ethanol and acidic buffers.
-
(viii)
After the dialysis, transfer the solution to an RNase-free 1.5 mL tube and measure the volume.
-
(ix)
Compensate the solution with 1× PBS to a final volume of 1,000 µL.
Pause point
The resulting solution can be stored at 4 °C for a few days before use. However, it is recommended that the formulated LNPs be used as soon as possible to maintain consistent results. Storage at RT is not recommended. Storage at freezing temperatures is also not recommended unless optimized cryoprotectants are used.
-
(i)
-
(C)
Preparation of the base 4A3-SC8 mDLNP formulation by the microfluidic mixing method
Timing 40 min plus at least 2 h for dialysis
Critical step
A protocol is described here to use the Benchtop NanoAssemblr. If using other Precision Nanosystems models, other microfluidic mixing equipment from other companies, or homemade microfluidic setups, then the steps will have to be adjusted following recommended protocols from manufacturers and literature reports.
-
(i)
Add 48.6 µL of the 4A3-SC8 mDLNP complete lipid mix solution (prepared in Box 1, step 8) into an RNase-free 1.5 mL tube.
-
(ii)
Add 251.4 µL ethanol into the above tube.
-
(iii)
Transfer the ethanol phase prepared from Step 26C(ii) to a 1 mL syringe. Remove the air bubbles.
-
(iv)
Take another RNase-free 1.5 mL tube, add 880 µL of citrate buffer (10 mM, pH 4) and then add 20 µL of Luc mRNA (1.0 mg/mL). Mix well.
Pause point
The mRNA concentration can be measured by NanoDrop.
-
(v)
Transfer the aqueous phase prepared from Step 26C(iv) into a 5 mL syringe. Remove the air bubbles.
-
(vi)
Set up the Nano-assembler platform. Insert the cartridge into the equipment. Load the two prefilled syringes. Load two collection tubes: one is for the sample, and the other is for the waste.
-
(vii)
Set the parameters on the software. Set the total injection volume as 1.0 mL and the total flow rate as 12 mL/min (aqueous phase loaded syringe dispenses 0.75 mL at a flow rate of 9 mL/min; ethanol phase loaded syringe dispenses 0.25 mL at a flow rate of 3 mL/min). Set the total waste volume as 0.40 mL (start waste is 0.35 mL, and end waste is 0.05 mL).
-
(viii)
Click ‘Start’ to make the formulation. Theoretically, there should be ~0.40 mL in the ‘waste’ tube and ~0.60 mL of formulated LNPs in the ‘sample’ tube.
Critical step
The volume ratio between the aqueous phase and ethanol phase is set to be 3:1. Use deionized water and ethanol to wash the cartridge after each formulation run. Overusing a cartridge may jeopardize experimental results. Cartridges can become blocked, which may cause loss of materials and damage to the instrument. Cartridge overuse or failure to wash cartridges also increases the risk of cross contamination.
-
(ix)
Incubate the resulting solution in the ‘sample’ collection tube at RT for up to 15 min.
Pause point
This incubation step is optional. It is recommended to perform the dialysis within 15 min after mixing.
-
(x)
Dialyze the above solution using a Pur-A-Lyzer Maxi 3500 Dialysis tube against 1× PBS for at least 2 h to remove the ethanol.
Critical step
Increase the dialysis time if more ethanol and acidic buffers are used.
-
(xi)
After the dialysis, transfer the solution to an RNase-free 1.5 mL tube and measure the volume.
-
(xii)
Compensate the solution with 1× PBS to a final volume of 1,000 µL.
Pause point
The resulting solution can be stored at 4 °C for a few days before use. However, it is recommended to use the formulated LNPs as soon as possible to maintain consistent results. Storage at RT is not recommended. Storage at freezing temperatures is also not recommended unless optimized cryoprotectants are used.
-
(i)
-
(A)
Part 4: characterization of SORT LNPs
-
27
The mRNA LNPs prepared as described above should be characterized before usage. The resulting data can verify proper LNP formulation. If deviations from expected results are observed, the resulting characterization data can be used to identify errors, interpret downstream, functional results and guide optimization. To determine the particle sizes of the LNPs, perform the steps in option A. To measure the encapsulation efficiency of mRNA in LNP formulations, follow the guidelines in option B. To use the LNPs in further in vitro and in vivo experiments, the anticipated particle sizes are <200 nm and the anticipated encapsulation efficiencies are >70%.
-
(A)
Measurement of LNP particle size by DLS
Timing 20 min
Critical
Taking 4A3-SC8 mDLNPs as a typical example, the concentration of mRNA in the 4A3-SC8 mDLNP formulation prepared in Step 26 is 0.01 mg/mL. If other formulations are made, or if concentrations are adjusted, then the following steps will need to be adjusted accordingly.
-
(i)
Pipette 10 µL of the resulting formulation from either pipette, vortex or microfluidic mixing in Step 26 into an RNase-free 1.5 mL tube.
-
(ii)
Add 90 µL of 1× PBS into the above tube for dilution.
-
(iii)
Transfer the diluted solution to a disposable cuvette.
-
(iv)
Insert the cuvette into a Zetasizer Nano ZS machine (Malvern, v.7.13) with a He-Ne laser (λ = 632 nm). Measure the particle size of the sample following manufacturer recommended settings.
-
(i)
-
(B)
Measurement of the encapsulation efficiency of mRNA in LNPs
Timing 1 h
Critical
The steps generally follow the Quant-iT RiboGreen recommended test protocols.
-
(i)
Prepare a 2 µg/mL solution of RNA in 1× TE: add 1 µL of the Luc mRNA (1.0 mg/mL) into 499 µL of 1× TE, and mix well.
-
(ii)
Prepare a 100 ng/mL stock solution of RNA in TE: dilute the 2 µg/mL RNA solution prepared in Step 27B(i) 20-fold into 1× TE.
-
(iii)
Prepare a 2,000-fold diluted Quant-iT RiboGreen reagent: dilute the Quant-iT RiboGreen RNA Reagent 2,000-fold with 1× TE.
Critical step
Protect the Quant-iT RiboGreen reagent from light by covering with foil or placing it in the dark to reduce photodegradation. For best results, use the diluted solution within a few hours of preparation.
-
(iv)
Prepare the RNA standard dilution series presented in Table 7.
Table 7 Protocol for preparing an RNA standard curve -
(v)
To prepare LNP formulations containing 100 ng/mL mRNA, dilute the mRNA LNPs prepared in Step 26 (0.01 mg/mL RNA concentration) 100-fold with 1× TE.
-
(vi)
To prepare LNP formulation dilutions with an mRNA concentration of 25 ng/mL, dilute the LNP formulations containing 100 ng/mL mRNA prepared from Step 27B(v) to a final RNA concentration of 25 ng/mL by adding 175 µL of LNP formulations containing 100 ng/mL RNA from Step 27B(v), 350 µL of 2,000-fold diluted Quant-iT RiboGreen reagent from Step 27B(iii), and 175 µL of 1× TE into a 1.5 mL RNAse-free tube. Mix them well.
-
(vii)
In a 96-well plate (black and flat bottom), add all the samples of RNA standard dilution series prepared in Step 27B(iv) and LNP formulation dilutions prepared in the previous step in triplicate with a final volume of 200 μL per well. Mix well, and incubate for 2–5 min at RT. Protect the plate from light.
-
(viii)
Measure the sample fluorescence using the microplate reader and standard fluorescein wavelengths (excitation ~485 nm, emission ~535 nm).
Critical step
Test the mRNA standard dilution series and LNP formulation dilutions in the same measurement.
-
(ix)
Subtract the fluorescence value of the blank (Table 7) from that of each sample. Use corrected data to generate a standard curve of fluorescence versus RNA concentration. Encapsulated mRNA within LNPs should be blocked from Ribogreen detection, allowing only free unencapsulated mRNA to be quantified.
-
(x)
Calculate the concentration of unencapsulated mRNA in solution (C, ng/mL) according to the standard curve.
-
(xi)
Calculate the encapsulation efficiency of mRNA in each formulation by: (1 − C/25) × 100 (%).
-
(i)
-
(A)
Part 5: in vitro delivery of Luc mRNA LNPs
Timing 2–3 d
Critical
Two representative mRNA delivery LNPs (4A3-SC8 mDLNPs and MC3 LNPs) prepared by the pipette mixing method in Step 26A can be diluted to formulations containing 1 ng/µL Luc mRNA and used for in vitro experiments. Follow the guidelines below. Lipofectamine RNAiMAX, which is a proprietary cationic lipid formulation, is used as a positive control.
-
28
Preseed 1 × 104 cells (IGROV-1 cells suspended in RPMI-1640 medium with 10% FBS) in each well of a white opaque 96-well plate, and incubate the cells at 37 °C in a humidified 5% CO2 atmosphere overnight.
-
29
Prepare 4A3-SC8 mDLNPs containing 1 ng/µL Luc mRNA by diluting the 4A3-SC8 mDLNPs prepared in Step 26A (containing 10 ng/µL) tenfold with 1× PBS.
-
30
Prepare MC3 LNPs containing 1 ng/µL Luc mRNA by diluting the MC3 LNPs prepared in Step 26A (containing 10 ng/µL) tenfold with 1× PBS.
-
31
Positive control. Prepare RNAiMAX formulations containing 1 ng/µL Luc mRNA by adding 1.5 µL of RNAiMAX reagent into 25 µL of Opti-MEM medium to form the diluted RNAiMAX reagent. Add 1 µL of Luc mRNA in 25 µL of Opti-MEM medium to form the diluted mRNA. Combine the diluted RNAiMAX reagent and diluted mRNA together, and incubate at RT for 5 min. Dilute the resulting formulation with PBS to obtain an mRNA concentration of 1 ng/µL.
-
32
Negative control. Dilute 1 mg/mL mRNA with 1× PBS to an mRNA concentration of 1 ng/µL. PBS and untreated wells can serve as additional negative controls.
-
33
Replace the old medium in each well of the plate from Step 28 with 150 µL of fresh medium. Add the same volume (25 µL per well) of each sample (4A3-SC8 mDLNPs, MC3 LNPs, negative control or positive control, from Steps 29–32, respectively) or fresh medium (blank) into five parallel wells, and incubate for 24 h.
-
34
After 24 h of incubation, gently remove and discard the medium by careful pipette or vacuum aspiration. Dilute the prepared AFC reagent fivefold with a serum-free RPMI-1640 medium. Add 50 µL of the diluted AFC solution into each well, and incubate at 37 °C for 30 min.
-
35
Measure the cell viability using a microplate reader. It is expected to see a cell viability >90%.
-
36
Add 25 µL of prepared ONE-Glo Luciferase Assay reagent into each well, mix briefly using microplate shaker and measure the luciferase expression using a microplate reader. The anticipated in vitro luciferase expression may reach as high as ~106 relative light units.
Part 6: in vivo delivery of Luc mRNA LNPs
Timing 1–2 d per group following acclimation of animals
Critical
C57BL/6 mice (female, 6–8 weeks old, ~20 g) were intravenously injected with various Luc mRNA formulations prepared by all three different methods in Step 26 (n = 3 per group) and imaged for luminescence by IVIS. The above protocols describe methods to achieve dispersed LNP concentrations in PBS suitable to administer 200 µL into 20 g mice via the tail vein.
Critical
For animals with different weights, or LNPs aimed to be dosed higher or lower than 0.1 mg/kg, the concentrations can be adjusted accordingly.
-
37
For one mouse (~20 g), intravenously inject 200 µL of the resulting Luc mRNA LNP from Step 26A(viii), 26B(ix) or 26C(xii) to achieve a 0.1 mg/kg mRNA injection dosage. Further note that, if the animal weighs much more than 20 g or less than 20 g, then the injection volume should be compensated to reach an injection dosage of 0.1 mg/kg. Wait for 6 h before IVIS measurement.
-
38
Set up the IVIS imaging system.
-
39
Anesthetize the animals using the rodent anesthesia system with isoflurane (2.5% (vol/vol) in 0.2 L/min O2 flow).
-
40
Intraperitoneally inject d-luciferin solution dissolved in 1× PBS into a mouse at a dose of 150 mg/kg. Scan the mouse with the IVIS imaging system 3 min after administration. IVIS settings: camera temperature −90 °C, stage temperature 37 °C, exposure time 60 s, binning ‘medium’, F/Stop ‘1’.
-
41
Euthanize the mice in a chamber with a CO2 fill rate of 30–70% of the chamber volume per minute. Dissect the mouse and collect the major organs (heart, lungs, liver, spleen and kidneys). Image and quantify the luciferase expression of the organs by the IVIS system.
Troubleshooting
Troubleshooting advice can be found in Table 8.
Timing
-
Steps 1–14, synthesis of AEMA: 1–2 d
-
Steps 15–25, synthesis of 4A3-SC8: 3–4 d
-
Step 26A, preparation of the base 4A3-SC8 mDLNP formulation by the pipette mixing method: 30 min for mixing plus at least 1 h for dialysis
-
Step 26B, preparation of the base 4A3-SC8 mDLNP formulation by the vortex mixing method: 30 min plus at least 1 h for dialysis
-
Step 26C, preparation of the base 4A3-SC8 mDLNP formulation by the microfluidic method: 40 min plus at least 2 h dialysis
-
Step 27A, measurement of LNP particle size: 20 min
-
Step 27B, measurement of the encapsulation efficiency of mRNA in LNPs: 1 h
-
Steps 28–36, in vitro delivery of Luc mRNA LNPs: 2–3 d
-
Steps 37–41, in vivo delivery of Luc mRNA LNPs: 1–2 d per group
-
Box 1, steps for making complete lipid mix solutions for 4A3-SC8 formulations in ethanol: 1–2 h
-
Box 2, steps for making complete lipid mix solutions for MC3 formulations in ethanol: 1–2 h
Anticipated results
Synthesis of AEMA (compound 1)
After Steps 12 and 13, a colorless oil-like compound should be obtained. 1H NMR spectrum of AEMA in CDCl3 is shown in Supplementary Fig. 1. 1H NMR (400 MHz, CDCl3): δ 6.38 (dd, J = 17.3, 1.5 Hz, 1H), 6.14–6.02 (m, 2H), 5.81 (dd, J = 10.4, 1.5 Hz, 1H), 5.54 (p, J = 1.6 Hz, 1H), 4.35 (m, J = 8.1, 6.4, 4.7, 3.2, 2.1 Hz, 4H), 1.94–1.80 (m, 3H) (J, coupling constant; dd, double of doublets; m, multiplet; p, pentet). MS: m/z calculated for C9H12O4 [M + H]+: 185.081; found: 185.080.
Synthesis of 4A3-SC8 (compound 2)
After Step 24, a colorless and sticky oil compound should be obtained. 1H NMR spectrum of 4A3-SC8 in CDCl3 is shown in Supplementary Fig. 2. 1H NMR spectrum (400 MHz, CDCl3): δ 4.40–4.21 (m, 16H), 2.90–2.64 (m, 16H), 2.64–2.39 (m, 24H), 2.30 (t, J = 7.4 Hz, 4H), 2.20 (s, 3H), 1.68–1.52 (m, 12H), 1.44–1.21 (m, 52H), 0.96–0.80 (m, 12H) (s, singlet; t, triplet). MS: m/z calculated for C75H139N3O16S4 [M + 2H]2+: 733.959; found: 733.956.
SORT LNP formulations prepared by three technical methods
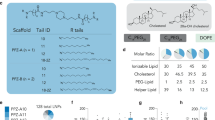
Results of RiboGreen assay (Fig. 4b) and DLS (Fig. 4c) measurements demonstrate high mRNA encapsulation and well-controlled sizes of 4A3-SC8-based LNPs (Fig. 4a) prepared by all three technical methods. Except for the 4A3-SC8 spleen SORT LNP with 10% 18PA (a negatively charged lipid that can repel negatively charged mRNA during self-assembly), the encapsulation efficiencies of mRNA for all other 4A3-SC8-based LNPs are >80% (Fig. 4b). Among them, formulation with 50% DOTAP has the highest encapsulation efficacy attributed to sufficient positive charges available for encapsulation of mRNA. The encapsulation efficiencies of LNPs prepared by the microfluidic mixing method are slightly higher than those of LNPs prepared by pipette and vortex mixing methods, which are in agreement with other reports on nucleic acid LNP systems44,51,52. DLS results reflect that 4A3-SC8-based LNPs prepared by the three different methods all have well-controlled structures. The intensity-weighted hydrodynamic distributions show a single peak with a low polydispersity index (PDI) <0.2. LNPs prepared by pipette and vortex mixing methods have larger sizes due to relatively slower mixing rates where mass transport effects may result in larger particles. However, the microfluidic mixing method generates LNPs with much smaller sizes (<100 nm). It is expected that, at a sufficiently fast mixing rate, LNPs formed by precipitation in response to the rapidly rising polarity of the environment will adopt a ‘limit size’ compatible with the physical properties of component molecules51. Similar phenomena are also observed for MC3-based LNPs (Fig. 5b–d).

a, Formulation details of base four-component 4A3-SC8 mDLNPs and five-component 4A3-SC8 SORT LNPs. b, Encapsulation efficiency of mRNA in 4A3-SC8-based LNPs. c, Representative dynamic light scattering (DLS) analysis of 4A3-SC8-based LNPs, particle sizes and PDIs are shown. d–g, 4A3-SC8-based Luc mRNA SORT LNPs were prepared by three mixing methods and injected intravenously into mice at a dosage of 0.1 mg/kg Luc mRNA. After 6 h, ex vivo images of luminescence (from top to bottom rows: pipette, vortex and microfluidic mixing methods) in major organs (heart, lungs, liver, spleen and kidneys) and quantification data are shown. Data are presented as mean ± s.e.m. (n = 3 biologically independent animals). 4A3-SC8 SORT LNPs with 20% DODAP (e) enhanced liver delivery. Inclusion of 50% DOTAP (f) and 10% 18PA (g) shifted the luciferase protein expression to the lungs and spleen, respectively. Statistical analysis of data in Fig. 4d–g is shown in Supplementary Fig. 4. Please see the Data Availability Statement and source data for more information. All the animal experiments were approved by the IACUC of the UTSW and were consistent with local, state and federal regulations as applicable.

a, Formulation details of base four-component MC3 LNPs and five-component MC3 SORT LNPs. b, Encapsulation efficiency of mRNA in MC3-based LNPs. c, Representative DLS analysis of MC3-based LNPs, particle sizes and PDIs are shown. d–f, MC3-based Luc mRNA SORT LNPs were prepared by three mixing methods and injected intravenously into mice at a dosage of 0.1 mg/kg Luc mRNA. After 6 h, ex vivo images of luminescence (from top to bottom rows: pipette, vortex and microfluidic methods) in major organs (heart, lungs, liver, spleen and kidneys) and quantification data are shown. Data are presented as mean ± s.e.m. (n = 3 biologically independent animals). MC3 LNPs mainly deliver mRNA to the liver. Inclusion of 50% DOTAP (e) and 30% 18PA (f) shifted the luciferase protein expression to the lungs and spleen, respectively. Statistical analysis of data in Fig. 5d–f are shown in Supplementary Fig. 4. Please see the Data Availability Statement and source data for more information. All the animal experiments were approved by the IACUC of the UTSW and were consistent with local, state, and federal regulations as applicable.
The base 4A3-SC8 mDLNPs and MC3 LNPs prepared by the pipette mixing method were used as representatives for evaluation of in vitro mRNA delivery. These two LNP systems both show negligible cytotoxicity (Supplementary Fig. 3a) and exhibit higher luciferase expression than the positive control RNAiMAX (Supplementary Fig. 3b).
Base 4A3-SC8 LNPs and 4A3-SC8 SORT LNPs (liver, lungs and spleen) were prepared by all three methods and administered intravenously into the tail veins of mice at a dosage of 0.1 mg/kg Luc mRNA. After 6 h, the resulting luciferase protein expression demonstrated that adding different SORT molecules alters the tissue tropism (Fig. 4d–g). Administration of 20% DODAP 4A3-SC8 LNP enhances liver delivery, and the luciferase expression improves several times compared with base mDLNPs. Incorporation of a permanently cationic SORT lipid (50% DOTAP) systematically shifts the luminescence activity to lungs, while inclusion of a permanently anionic SORT lipid (10% 18PA) aids spleen delivery, albeit with some reduction in overall activity. We note that the SORT methodology is also generally applicable to MC3-based LNPs, wherein formulating MC3 LNPs with 50% DOTAP and 30% 18PA alters the protein expression profile from the liver to lungs to spleen (Fig. 5d–f), mirroring the results of 4A3-SC8 SORT LNPs. Our previous study indicated that 5–40% 18PA incorporation resulted in exclusive mRNA delivery to the spleen. Fine tuning this percentage, we note here that 30% 18PA is preferable for spleen delivery in MC3 system, while the molar ratio should be reduced to 10% for making preferable 4A3-SC8 spleen SORT LNPs, reflecting the fact that the percentage of SORT molecule is of crucial importance for tissue-specific delivery with sufficiently high mRNA encapsulation. We used three different technical methods to prepare the SORT LNP formulations. The results indicate that the formulation techniques do not affect the utilization of SORT and the size of the LNPs did not substantially affect the tissue tropism. The luciferase expressions of LNPs prepared by the pipette and vortex mixing methods are on the same level. Size did slightly affect efficacy, which was also observed in our previous paper24. Except for 10% 18PA 4A3-SC8 LNPs, all other SORT LNPs prepared by the microfluidic method possess a slightly higher luciferase activity, especially for 50% DOTAP formulation. However, the luciferase expression decreases when formulated with 10% 18PA by microfluidic method, which might be attributed to charge repulsion in the herringbone structures of the cartridge. The above results are more obvious in MC3-based LNPs. Overall, these three technical methods to prepare SORT LNPs are comparable, and one can make appropriate selection depending on the desired goals and applications. There was consistency across the batches in terms of physical properties and in vitro/in vivo results.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting this protocol are available within the article and its Supplementary Information files and from the corresponding author upon reasonable request.
References
Sahin, U., Karikó, K. & Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780 (2014).
Hajj, K. A. & Whitehead, K. A. Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2, 17056 (2017).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2020).
Hou, X., Zaks, T., Langer, R. & Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019).
Li, B., Zhang, X. & Dong, Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotech. 11, e1530 (2019).
Wei, T. et al. Delivery of tissue-targeted scalpels: opportunities and challenges for in vivo CRISPR/Cas-based genome editing. ACS Nano 14, 9243–9262 (2020).
Kulkarni, J. A. et al. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16, 630–643 (2021).
Allen, T. M. & Cullis, P. R. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48 (2013).
Alameh, M.-G. et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892 (2021).
Elia, U. et al. Design of SARS-CoV-2 hFc-conjugated receptor-binding domain mRNA vaccine delivered via lipid nanoparticles. ACS Nano 15, 9627–9637 (2021).
Hassett, K. J. et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 15, 1–11 (2019).
McKay, P. F. et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 11, 3523 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
VanBlargan, L. A. et al. An mRNA vaccine protects mice against multiple tick-transmitted flavivirus infections. Cell Rep. 25, 3382–3392.e3383 (2018).
Li, B. et al. An orthogonal array optimization of lipid-like nanoparticles for mRNA delivery in vivo. Nano Lett. 15, 8099–8107 (2015).
Robinson, E. et al. Lipid nanoparticle-delivered chemically modified mRNA restores chloride secretion in cystic fibrosis. Mol. Ther. 26, 2034–2046 (2018).
Veiga, N. et al. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 9, 4493 (2018).
Cheng, Q. et al. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 30, 1805308 (2018).
Patel, S. et al. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 303, 91–100 (2019).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
Han Jeong, P. et al. In vivo delivery of CRISPR–Cas9 using lipid nanoparticles enables antithrombin gene editing for sustainable hemophilia A and B therapy. Sci. Adv. 8, eabj6901 (2022).
Liu, S. et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat. Mater. 20, 701–710 (2021).
Qiu, M. et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl Acad. Sci. USA 118, e2020401118 (2021).
Wei, T. et al. Systemic nanoparticle delivery of CRISPR–Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 11, 3232 (2020).
Billingsley, M. M. et al. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 20, 1578–1589 (2020).
Lee, K. et al. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 8, 1101–1105 (2020).
Miao, L. et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 37, 1174–1185 (2019).
Oberli, M. A. et al. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 17, 1326–1335 (2017).
Sahin, U. et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585, 107–112 (2020).
Zhang, H. et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl Acad. Sci. USA 118, e2005191118 (2021).
Li, W. & Szoka, F. C. Lipid-based nanoparticles for nucleic acid delivery. Pharm. Res. 24, 438–449 (2007).
Cheng, X. & Lee, R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 99, 129–137 (2016).
Jokerst, J. V., Lobovkina, T., Zare, R. N. & Gambhir, S. S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 6, 715–728 (2011).
Akinc, A. et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 14, 1084–1087 (2019).
Lee, S. M. et al. A systematic study of unsaturation in lipid nanoparticles leads to improved mRNA transfection in vivo. Angew. Chem. Int. Ed. 60, 5848–5853 (2021).
Álvarez-Benedicto, E. et al. Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA). Biomater. Sci. 10, 549–559 (2022).
Farbiak, L. et al. All-in-one dendrimer-based lipid nanoparticles enable precise HDR-mediated gene editing in vivo. Adv. Mater. 33, 2006619 (2021).
Dilliard, S. A., Cheng, Q. & Siegwart, D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl Acad. Sci. USA 118, e2109256118 (2021).
Zhou, K. et al. Modular degradable dendrimers enable small RNAs to extend survival in an aggressive liver cancer model. Proc. Natl Acad. Sci. USA 201520756 (2016).
Stroock Abraham, D. et al. Chaotic mixer for microchannels. Science 295, 647–651 (2002).
Chen, D. et al. Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J. Am. Chem. Soc. 134, 6948–6951 (2012).
Belliveau, N. M. et al. Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol. Ther. Nucleic Acids 1, e37 (2012).
Swingle, K. L. et al. Amniotic fluid stabilized lipid nanoparticles for in utero intra-amniotic mRNA delivery. J. Control. Release 341, 616–633 (2022).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
Hirai, Y. et al. Charge-reversible lipid derivative: a novel type of pH-responsive lipid for nanoparticle-mediated siRNA delivery. Int. J. Pharm. 585, 119479 (2020).
Maeta, M. et al. Vitamin E scaffolds of pH-responsive lipid nanoparticles as DNA vaccines in cancer and protozoan infection. Mol. Pharm. 17, 1237–1247 (2020).
Evers, M. J. W. et al. State-of-the-art design and rapid-mixing production techniques of lipid nanoparticles for nucleic acid delivery. Small Methods 2, 1700375 (2018).
Meure, L. A., Foster, N. R. & Dehghani, F. Conventional and dense gas techniques for the production of liposomes: a review. AAPS PharmSciTech 9, 798 (2008).
Leung, A. K. K. et al. Microfluidic mixing: a general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B 119, 8698–8706 (2015).
Shepherd, S. J., Issadore, D. & Mitchell, M. J. Microfluidic formulation of nanoparticles for biomedical applications. Biomaterials 274, 120826 (2021).
Zhou, K. et al. Hydrophobic domain structure of linear-dendritic poly(ethylene glycol) lipids affects RNA delivery of lipid nanoparticles. Mol. Pharm. 17, 1575–1585 (2020).
Acknowledgements
D.J.S. acknowledges financial support from the National Institutes of Health (NIH) National Institute of Biomedical Imaging and Bioengineering (NIBIB) (R01 EB025192-01A1) and National Cancer Institute (R01 CA269787-01), the Cystic Fibrosis Foundation (CFF) (SIEGWA18XX0 and SIEGWA21XX0) and the Cancer Prevention and Research Institute of Texas (CPRIT) (RP190251). S.M.L. and J.R. acknowledge support from the NIH (T32GM127216). T.W. acknowledges support from a CPRIT Training Grant (RP160157). The authors acknowledge the UTSW Small Animal Imaging Shared Resource, which is supported in part by the NIH NCI Support Grant (P30 CA142543).
Author information
Authors and Affiliations
Contributions
X.W. and D.J.S. conceived and designed the experiments. X.W. performed the majority of experiments, analyzed the data and wrote the first draft of the manuscript. S.L. assisted with the design of the experiments, chemical synthesis, in vitro and in vivo studies. Y.S. and X.Y. assisted in improving mixing methods for the preparation of LNP formulations. X.Y., S.M.L., J.G. and J.R. assisted with the chemical synthesis. Q.C. and T.W. assisted with the design of the experiments. D.Z. assisted with the vortex mixing method. X.L. and P.B. assisted with the preparation of complete lipid mix solutions and general experiments. D.J.S. supervised all the experiments, wrote and edited the manuscript, and managed the project. All authors contributed to the development of the protocol and improved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The University of Texas System has filed patent applications related to the SORT technology, with some authors listed as co-inventors. D.J.S. is a co-founder/consultant to ReCode Therapeutics, which has licensed intellectual property from UT Southwestern. D.J.S. is on the Scientific Advisory Board of Tome Biosciences.
Peer review
Peer review information
Nature Protocols thanks Michael Mitchell, Dan Peer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Cheng, Q. et al. Nat. Nanotechnol. 15, 313–320 (2020): https://doi.org/10.1038/s41565-020-0669-6
Wei, T. et al. Nat. Commun. 11, 3232 (2020): https://doi.org/10.1038/s41467-020-17029-3
Dilliard, S. A. et al. Proc. Natl. Acad. Sci. USA 118, e2109256118 (2021): https://doi.org/10.1073/pnas.2109256118
Supplementary information
Supplementary Information
Supplementary Figs. 1–4.
Supplementary Data
Statistical data for Supplementary Fig. 3.
Source data
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, X., Liu, S., Sun, Y. et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat Protoc 18, 265–291 (2023). https://doi.org/10.1038/s41596-022-00755-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-022-00755-x
This article is cited by
-
Strategies to reduce the risks of mRNA drug and vaccine toxicity
Nature Reviews Drug Discovery (2024)
-
RNAi-based drug design: considerations and future directions
Nature Reviews Drug Discovery (2024)
-
Gene targeting in adult organs using in vivo cleavable donor plasmids for CRISPR-Cas9 and CRISPR-Cas12a
Scientific Reports (2024)
-
The therapeutic potential of immunoengineering for systemic autoimmunity
Nature Reviews Rheumatology (2024)
-
Targeting strategies with lipid vectors for nucleic acid supplementation therapy in Fabry disease: a systematic review
Drug Delivery and Translational Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
