
Abstract
Macrophages in atherosclerotic lesions promote plaque progression and are an attractive therapeutic target in cardiovascular research. Here we present a protocol for synthesis of small interfering RNA (siRNA) nanoparticles (NP) that target lesional macrophages as a potential treatment for atherosclerosis. Ca2+/calmodulin-dependent protein kinase γ (CaMKIIγ) activity in macrophages of advanced human and mouse atherosclerotic plaques drives necrosis by downregulating the expression of the efferocytosis receptor MerTK. Therefore, selective inhibition of CaMKIIγ in lesional macrophages holds great promise for the treatment of advanced atherosclerosis. We recently developed a siRNA NP platform that can selectively silence CaMKIIγ in macrophages, resulting in increased plaque stability. We provide a detailed protocol for the synthesis of NP components, the preparation and characterization (physicochemical and in vitro) of siRNA NPs, and the evaluation of in vivo therapeutic effects of siRNA NPs and their biocompatibility in atherosclerotic mice. Our siRNA-loaded polymer–lipid hybrid NPs are constructed via a robust self-assembly method, exhibiting excellent in vivo features for systemic siRNA delivery. Following this protocol, it takes 3–5 d to prepare the siRNA NPs, 8–10 d to characterize the NPs and 4–5 weeks to evaluate their therapeutic effects in established atherosclerotic mice. By changing the RNA molecules loaded in the NPs, lesional macrophages can be targeted for the exploration and validation of new targets/pathways in atherosclerosis.
Similar content being viewed by others

Introduction
Macrophage activation has been implicated in the pathogenesis of many human diseases, including atherosclerosis, obesity, diabetes, cancer and skin diseases1. Among them, atherosclerosis, an inflammatory disease involving arterial plaque deposition, which increases the risk of cardiovascular disease and stroke, is highly correlated with macrophage activation2,3. Therefore, inhibiting the activation of pro-atherogenic pathways in macrophages holds great promise for the treatment of atherosclerotic vascular diseases4. However, to date, only suboptimal therapeutic effects have been achieved due to the lack of therapeutics or treatments with high specificity and efficacy.
RNA interference (RNAi) is a powerful method capable of selectively silencing any gene of interest, showing great potential to treat various diseases5,6,7,8,9,10. The success of RNAi therapy largely depends on the efficient in vivo delivery of therapeutic small interfering RNA (siRNA) to the target cells. To reach the RNAi machinery in the cytoplasm, siRNA molecules need to overcome several physiological barriers, including being degraded by nucleases, being rapidly cleared by the mononuclear phagocyte system or renal filtration11, poor cellular uptake and inefficient endosomal escape8. Although the rapid development of siRNA technology has yielded relatively stable siRNA with reduced side effects through chemical modification and sequence optimization12,13,14, the efficient delivery of siRNA into target cells is still challenging. Therefore, it is highly desirable to develop a safe and efficient delivery vehicle that can protect the siRNA from enzymatic degradation, deliver it to target cells and facilitate its cellular uptake and endosomal escape.
To this end, various delivery systems have been developed for more effective delivery of siRNA15. Many of them have been investigated in clinical trials for the treatment of different diseases, including cancer, genetic disorders, fibrosis, hypercholesterolemia and viral infections16. The research interests of the scientific community in siRNA-based therapy have been fueled by the first US Food and Drug Administration-approved nano-formulated siRNA drug (Patisiran, Alnylam Pharmaceuticals), treating a rare genetic disorder17. Although several polymeric or lipid nanoparticle (NP)-based siRNA delivery systems have been developed for the treatment of vascular diseases18,19,20,21,22,23,24, siRNA-based therapy via targeting macrophages in atherosclerotic lesions for atherosclerosis treatment is still in its infancy. It is worth mentioning that Leuschner et al. pioneered the siRNA-based therapy using cationic lipid NPs for in vivo treatment of atherosclerosis via targeting macrophages25. While these polymeric or lipid NP-based systems have shown satisfying therapeutic effects, they might not be applicable in the treatments where higher gene therapy efficacy is required. Therefore, novel strategies that can further enhance the protection of siRNA and prolong its in vivo circulation time are needed.
Development of the protocol
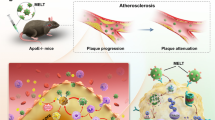
To address outstanding limitations in nucleic acid delivery to macrophages, we developed a macrophage-targeting siRNA NP platform for the treatment of atherosclerosis via selectively silencing a functional gene in macrophages of atherosclerotic lesions26. The macrophage gene Camk2g, encoding a calcium-activated kinase named CaMKIIγ (Ca2+/calmodulin-dependent protein kinase γ), was chosen as the target based on our previous work. We demonstrated that the activation of lesional macrophage CaMKIIγ in advanced human and mouse atherosclerotic plaques was a major stimulus for the formation of necrotic lesions covered with thin fibrous caps27. Efferocytosis is a specialized phagocytic process for the removal of dead cells28. Mechanistic studies revealed that the CaMKIIγ suppresses the activation of transcription factor 6 (ATF6)/liver X receptor alpha (LXRα)/c-Mer proto-oncogene tyrosine kinase (MerTK) pathway that usually promotes the efferocytosis. Consequently, the inefficient efferocytosis causes secondary necrosis of uncleared cells, which eventually results in plaque necrosis and fibrous cap thinning (Fig. 1)27. Thin-capped, necrotic lesions destabilize the plaques in humans, leading to harmful consequences such as plaque rupture, acute luminal thrombosis and various acute atherothrombotic vascular events29.

The introduction of siCamk2g into the macrophages silences CaMKIIγ, resulting in the activation of the MerTK pathway, which further enhances efferocytosis. The increased efferocytosis leads to a decrease in plaque necrosis and fibrous cap thinning, which synergistically improve the plaque stability26,27.
Our siRNA NPs are rationally designed and constructed. First, a cationic lipid is used to complex the siRNA via electrostatic interactions. Then, a hydrophobic poly(lactic-co-glycolic) acid (PLGA) polymer further interacts with the siRNA/cationic lipid complex via hydrophobic interactions, followed by PEGylation of the complex using lipid–polyethylene glycol (lipid–PEG), yielding the siRNA NPs. To enable the targeted delivery of siRNA to macrophages, the macrophage-targeting peptide ligand S2P is conjugated to the siRNA NPs through the lipid–PEG. In a fat-fed Ldlr−/− mouse model with established atherosclerosis, the administration of the S2P-siCamk2g NPs can silence the CaMKIIγ of the lesional macrophage and subsequently enhance efferocytosis, resulting in thicker fibrous cap and less atherosclerotic plaque necrosis. Through this formulation, we developed a promising siRNA NP platform for the treatment of atherosclerosis via genetic manipulation of a specific molecule in lesional macrophages, providing a new therapeutic strategy for atherosclerosis and other related vascular diseases.
Here we outline the procedures for the preparation and characterization of siRNA NPs, and the evaluation of in vivo therapeutic effects of siRNA NPs in atherosclerotic mice. The NP components cationic lipid (G0-C14) and peptide-modified lipid–PEG (DSPE–PEG–S2P) can be easily synthesized by a one-step reaction. The siRNA-loaded polymer–lipid hybrid NPs are conveniently achieved by dropwise addition of the siRNA complex into the lipid–PEG aqueous solution at room temperature (25 °C). After quick purification by ultrafiltration, the resulting siRNA NPs can be readily used for in vitro and in vivo evaluation of gene silencing efficiency and therapeutic effects for atherosclerosis. We anticipate that these procedures will be instrumental for researchers seeking to build siRNA delivery systems to treat atherosclerosis and other diseases.
Overview of the procedure
The activation of lesional macrophages is one of the causes of the instability of the atherosclerotic plaques. We present here a robust strategy for the treatment of atherosclerosis in mice using lesional macrophage-targeting siRNA NPs. This protocol contains five main stages (Fig. 2). We first describe the synthesis and purification of NP components, including cationic lipid (G0-C14) and targeted lipid–PEG (DSPE–PEG–S2P) (Fig. 2a). Targeting is achieved by modifying the lipid with a peptide sequence that binds to/interacts with the macrophage receptor stabilin-2. Nuclear magnetic resonance (NMR) is used to confirm the structure of the compounds. We then provide a step-by-step nano-precipitation approach for the preparation of targeted siRNA NPs (Fig. 2b). The preparation and optimization of the macrophage-targeting siRNA NPs are key features of the protocol. Five components, including the synthesized (G0-C14, DSPE–PEG–S2P) and commercially available (siRNA, PLGA, DSPE–PEG) compounds, are used to construct the siRNA NPs. The ratio of components, the formulation, the stirring speed and the incubation time notably impact the properties of the resulting NPs. For example, if the stirring speed is much lower than the recommended speed (1,000 rpm), large NPs with low siRNA encapsulation efficiency (EE) will be formed. All the optimized parameters are discussed in this protocol. To confirm the successful formation of siRNA NPs, their physicochemical properties are fully characterized (Fig. 2c). Dynamic light scattering (DLS) is used to determine the size and colloidal stability of siRNA NPs. Transmission electron microscopy (TEM) is employed to characterize the morphology of siRNA NPs. Gel electrophoresis is used to investigate the stability of siRNA encapsulated by NPs. The resulting siRNA NPs are further characterized in vitro (Fig. 2d). Confocal microscopy is used to evaluate the endosomal escape ability of siRNA NPs by measuring the fluorescent signals of dye-labeled siRNA. A plate reader is implemented to quantify the gene silencing efficiency of the siRNA targeting luciferase (siLuc) NPs by measuring the bioluminescence signals. A plate reader is also used to assess the in vitro toxicity of siRNA NPs (the alamarBlue cell viability reagent). Finally, we evaluate the in vivo therapeutic effect of siCamk2g NPs in a WD-fed Ldlr−/− mouse model with established atherosclerosis (Fig. 2e). To confirm that the siCamk2g NPs can improve the plaque stability, microscopy is employed to determine the fibrous cap thickness and necrotic core area (picrosirius red staining). To further demonstrate that the enhanced plaque stability is ascribed to the promoted efferocytosis, fluorescence microscopy is implemented to characterize the macrophage-associated apoptotic cells (TUNEL assay). In addition, we evaluate the in vivo toxicity of the siRNA NPs using H&E staining and hematological analysis. Throughout the procedure (Fig. 2), we discuss the parameters that need to be optimized and how they influence the properties of the siRNA NPs, and how to troubleshoot any problems encountered when following the procedures.

a–e, The protocol comprises five main stages, including: synthesis of components (Steps 1–24) (a); preparation of the targeted siRNA NPs (Steps 25–52) (b); physicochemical characterization of the siRNA NPs (Steps 53–108) (c); in vitro characterization of the siRNA NPs (Steps 109–164) (d); and in vivo therapeutic effect evaluation of the siCamk2g NPs (Steps 165–189) (e).
Applications of the method
The key features of this protocol are the preparation, optimization and characterization of the siRNA-loaded polymer–lipid hybrid NPs, which have various applications in the biomedical field. The first application of this siRNA NP platform is the treatment of atherosclerosis, which is described in this protocol using a mouse model. It has been previously established that the activation of lesional macrophage CaMKIIγ in advanced human and mouse atherosclerotic plaques is a major stimulus for the formation of necrotic lesions covered with thin fibrous caps27. Therefore, the macrophage gene Camk2g, encoding CaMKIIγ, was chosen as the target for RNAi therapy. After administration to the mouse model with atherosclerosis, the targeted S2P-siCamk2g NPs were able to selectively silence the CaMKIIγ of the lesional macrophage and subsequently enhance efferocytosis, resulting in thicker fibrous cap and less atherosclerotic plaque necrosis26. These siRNA NPs hold great promise for the treatment of atherosclerosis as demonstrated in a mouse model. For the treatment of other diseases, potential users can obtain tailored siRNA NP platforms conveniently by replacing the siRNA used in this protocol with another functional siRNA sequence. For example, this siRNA NP platform has also been used successfully for the treatment of non-small cell lung cancer in a mouse model as demonstrated by another study in our group, in which Prohibitin 1 (PHB1) was chosen as the target to silence. The siPHB1-loaded NPs were <100 nm in diameter, had long blood circulation time (t1/2 of ~8.1 h), had high tumor accumulation (~5.2 mg/kg) and could effectively inhibit tumor growth in mice bearing NCI-H460 and A549 tumors6. In addition to siRNA delivery, the present NP platform can also be used for the delivery of large nucleic acid molecules such as mRNA with proper optimization30,31.
The wide deployment of two highly effective NP-based mRNA coronavirus disease 2019 (COVID-19) vaccines (mRNA-1273, Moderna32; and BNT162b2, Pfizer33) has fueled the research interests in NP-based mRNA delivery34,35,36,37. It is worth noting that the NPs used in these two vaccines are chemically dissimilar to our NPs. For example, they contain ionizable lipids rather than cationic lipids, and they do not contain PLGA or targeting ligands. Despite that, the application of our polymer–lipid hybrid NP platform for mRNA delivery for cancer therapy has been demonstrated in mouse models in two other studies by our group30,31. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a well-defined tumor suppressor gene that is lost or mutated in various human cancers38. The restoration of functional PTEN in PTEN-null tumor cells has been shown to suppress, at least temporarily, the growth of tumors. Indeed, the PTEN-mRNA can be efficiently incorporated into our polymer–lipid hybrid NPs with slight optimization to the protocol presented here. The resulting PTEN-mRNA-loaded NPs exhibit good stability in physiological environments and achieve high PTEN-mRNA transfection efficiency in prostate cancer cells without eliciting obvious cytotoxicity. Furthermore, in serval mouse models of prostate cancer, the systemic administration of the PTEN-mRNA-loaded NPs effectively inhibited the tumor growth in the mice30. Similarly, another tumor suppressor gene named p53 can also be employed as the therapeutic target for cancer therapy. We demonstrated that p53-mRNA-loaded polymer–lipid hybrid NPs can inhibit the tumor growth of mice bearing different tumor models, and also sensitize the tumors to mTOR inhibition31. Several recently published reviews have discussed the potential of our polymer–lipid hybrid NPs as an efficient siRNA or mRNA delivery platform for the treatment of different diseases39,40,41,42,43,44.
Overall, the polymer–lipid hybrid NP platforms generated by following our protocol are able to efficiently deliver siRNA and mRNA to target sites in vivo for the treatments of atherosclerosis and cancer, as validated by the studies of our group6,26,30,31. The NPs we prepared here have been modified with the targeting peptide S2P, a peptide that recognizes the macrophage receptor stabilin-2, allowing for the specific targeting of lesional macrophages. Besides the application for the treatment of atherosclerosis, our polymer–lipid hybrid NPs without any targeting ligand have been reported to show substantial accumulation in tumors, holding great promise for cancer therapy6. Modifications may be required before these NPs can be used for other applications. We anticipate that this protocol will be of interest to researchers who are developing RNA NPs to treat atherosclerosis, cancer and beyond. The techniques involved in this protocol can also be used by specialists in the fields of chemistry, nanotechnology, cardiovascular diseases, biology, RNA therapeutics and pharmacy.
Comparison with other methods
The present protocol describes the construction of a siRNA-loaded polymer–lipid hybrid NP platform for the treatment of atherosclerosis in mice via the regulation of lesional macrophage activation. Although our siRNA NPs-based therapeutic strategy for atherosclerosis has been rarely reported, a variety of other siRNA NP platforms have been developed for the treatment of a wide range of diseases15. One of the widely investigated siRNA NP platforms is the polymeric NP. For example, a PEGylated poly(guanidinium) (PEG-pGu)-based siRNA NP platform has been developed for Alzheimer’s disease therapy45. To increase the blood–brain barrier penetration, a galactose (Gal) ligand was introduced to the siRNA NPs. In a transgenic Alzheimer’s disease mouse model, the administrated Gal-siRNA NPs were able to cross the blood–brain barrier via glycemia-controlled glucose-transporter-1-mediated transport, improving the cognitive behavior of the mice. One concern of this cationic polymer system is the potential toxicity. To address this issue, a degradable influenza virus-inspired cationic polymer system was prepared for siRNA delivery46. Upon internalization by the cells, the resulting siRNA NPs could quickly escape from the endosome through an influenza virus mimicking endosomal escape mechanism, releasing the siRNA into the cytosol via the degradation of the polymer. Despite the fact that the toxicity issues of cationic polymers can be alleviated by the incorporation of degradable bonds, the instability of the polymeric NPs can be problematic when administrated in vivo. The only driving force for the siRNA/cationic polymer complex is electrostatic interactions, which can be largely weakened in the ionic strength condition of the body.
Another siRNA NP delivery platform is the DNA nanostructure47,48. As one of the well-characterized DNA nanostructures, DNA tetrahedron has been widely used for the delivery of therapeutics including siRNA. For instance, a self-assembled DNA tetrahedron was prepared for in vivo siRNA delivery49. The siRNA cargos were conveniently incorporated onto one of the six edges of the DNA tetrahedrons through one-step self-assembly. Importantly, different cancer-targeting ligands could be easily conjugated to surfaces of the DNA tetrahedrons via DNA hybridization. The high programmability of the DNA strands allowed the precise control of the size of the tetrahedrons, spatial orientation and density of the ligands. The siRNA-loaded DNA tetrahedral NPs could deliver the therapeutic siRNA to the tumor and silence the target genes. A similar siRNA-loaded DNA tetrahedral NP platform has also been developed for targeted delivery of therapeutic siRNA to the kidney of the mice, treating acute kidney injury50. In addition, a DNA nanosuitcase platform51 and a DNA nanotube platform52 have also been constructed for siRNA delivery. A major advantage of all these DNA nanostructure-based NP platforms is their negligible toxicity due to the inherent biocompatibility of DNA materials. However, these DNA nanostructures do not provide enough protection for the siRNA cargos, making them vulnerable to enzymatic degradation.
Among all the siRNA NP platforms, the lipid NP is the most clinically advanced platform. Indeed, the first US Food and Drug Administration-approved siRNA therapeutic (Patisiran) is based on lipid NP formulations17. This type of lipid NP is typically composed of: the ionizable lipid, phospholipid, cholesterol, lipid–PEG and siRNA53. Homogeneous lipid NPs with a diameter ~50–100 nm can be formed by rapidly mixing siRNAs with other components using a microfluidic mixing technique54. Notably, these lipid NPs are neutral at pH 7.4, but protonated in the acidic environment of endosomes, improving the endosomal escape of siRNA via the fusion of lipids with the membrane of endosomes. Despite the successful applications of siRNA lipid NPs55,56, specific designs might be required to improve their delivery efficiency for different diseases57. For example, for specific cell-type-related diseases such as macrophage-related atherosclerosis, a macrophage-targeting ligand can be introduced to the NPs to improve their therapeutic efficiency. Recently, a well-designed selective organ targeting NP platform showed its capability to specifically deliver therapeutics (Cas9 mRNA, single guide RNA) to lung, spleen and liver to enable efficient gene editing, holding great promise for the treatment of diverse organ-related diseases58,59.
Here, we provide a detailed protocol for the development of a polymer–lipid hybrid NP platform for efficient in vivo delivery of siRNA6,26. To date, siRNA-loaded polymer–lipid hybrid NPs have been typically synthesized by solvent evaporation or double emulsion methods60,61,62,63. However, our siRNA-loaded polymer–lipid hybrid NP is prepared through a simple nano-precipitation method, and has a unique structure consisting of a siRNA/cationic lipid/PLGA polymer core and a lipid–PEG shell. Compared with the ionizable lipid NP platform, we have introduced an extra hydrophobic polymer core to our platform, aiming to further protect the unstable nucleic acid cargo and prolong the blood circulation time. We have previously prepared different polymer–lipid hybrid NPs with several different cationic lipids, and the most efficient NP was prepared with G0-C14 lipid, which contains five tertiary amines and seven hydrocarbon tails6. We use the lowest concentration of G0-C14 (siRNA/G0-C14 weight ratio,1:15) that is able to condense the siRNA to prepare our siRNA NPs, while no pronounced in vitro and in vivo toxicities of the NPs are observed. The tertiary amines can provide sufficient positive charges to condense the negatively charged nucleic acids, and the hydrocarbon tails can bind to the hydrophobic polymer via hydrophobic interaction. Therefore, the amines and hydrocarbon tails are pivotal for the construction of our platform, whereas ionizable lipids do not have sufficient cationic charge to be suitable for the platform presented here.
Our polymer–lipid hybrid NP platform has several advantages over the other in vivo delivery platform discussed above. First, the NPs have a long blood circulation time (t1/2 of ~8.1 h compared with t1/2 of ~4 h for a MC3-based lipid NP64). The formation of the NPs is driven by the electrostatic interactions between the anionic nucleic acids and cationic lipids, and the hydrophobic interaction between the hydrophobic PLGA and lipid tails. Utilizing both electrostatic and hydrophobic forces produces NPs with high in vivo stability6. Second, targeting ligands can be easily introduced to the NPs. Given the multicomponent structure of the NPs, different targeting ligands can be conveniently introduced to the lipid–PEG shell without changing the main structure of the NPs. The ligand modification is described in this protocol26. In addition, to realize the on-demand release of the therapeutics, the hydrophobic PLGA polymer of the NPs can be replaced by a stimuli-responsive hydrophobic polymer. For example, we developed a redox-responsive NP platform by replacing the PLGA polymer with a reduction-responsive polymer31. Taken together, our polymer–lipid hybrid NP platform is highly flexible (to be engineered with different functions) and easy to prepare, holding great promise for efficient delivery of siRNA and other types of nucleic acids.
Experimental design
Here we describe the design and rationale for each procedure in this protocol.
Synthesis of cationic lipid and ligand (Steps 1–24)
The cationic lipid termed G0-C14 is synthesized by a unique synthetic strategy involving effective ring-opening of 1,2-epoxytetradecane (epoxide) by amine compound (poly (amido amide) (PAMAM) dendrimer G0) (Fig. 3a)60. This synthetic strategy requires no solvent and protection/deprotection steps, making it highly suitable for high-throughput screening of cationic lipids for superior nucleic acid delivery65. The generation 0 of PAMAM dendrimer is employed for the synthesis of cationic lipids based on its capability to condense the siRNA and its lower cytotoxicity than higher generations. In addition, we have previously synthesized cationic lipids using different cationic amino molecules, including G0, branched polyethylenimine (molecular weight ~800 g/mol), polypropylenimine tetramine dendrimer, generation 1 and diethylene triamine. Among them, the G0 lipid shows the highest siRNA delivery efficiency6. We chose 1,2-epoxytetradecane for the synthesis of cationic lipid because lipid-like compounds with 14 carbons have been shown to be optimal for siRNA delivery65. The maximal amounts of lipid tails that can be conjugated to G0 are eight according to the four free amines of G0. To obtain G0-C14 with one less tail than the fully conjugated G0-C14, seven equivalents (eq.) of lipid tails (1,2-epoxytetradecane) are reacted with one eq. of G0. The obtained crude product can be purified by silica gel chromatography with gradient elution (CH2Cl2 → 75:22:3 CH2Cl2/MeOH/NH4OH). The chemical structure of the G0-C14 can be determined by 1H NMR (the purified G0-C14 contains about seven lipid tails as calculated from the 1H NMR spectrum60).

The ligand-modified lipid–PEG (DSPE–PEG–S2P) is prepared by an efficient and widely used click reaction between the thiol group of the targeting peptide sequence: CRTLTVRKC (S2P) and the maleimide (Mal) group of Mal-modified DSPE–PEG (DSPE–PEG–Mal) (Fig. 3b). We use thiol–maleimide click chemistry for two reasons. First, the thiol group can be easily and conveniently introduced during the custom synthesis of the peptide. Hence, the peptide ligand can be conjugated to the lipid–PEG in one step. Second, the thiol and maleimide groups are stable in both organic and aqueous solutions, leading to high reaction efficiency. The resulting crude DSPE–PEG–S2P can be purified through washes with cold methanol (MeOH). The successful conjugation of S2P to DSPE–PEG can be validated by 1H NMR. If a user wants to target another cell type or organ, we recommend using an alternative cysteine-terminated peptide or other targeting ligand in place of S2P.
Preparation of nucleic acid-loaded NPs (Steps 25–52)
The siRNA NPs are prepared by a robust nano-precipitation method that is well established by our group. Due to the vulnerability of free siRNA to nucleases, the siRNA needs to be fully protected before it reaches its intended destination. To this end, our NPs are designed to have a core-shell structure, where the siRNA is loaded in the core and protected from enzymatic degradation. To fabricate the siRNA-containing core, we first condense the siRNA with cationic lipid G0-C14 through electrostatic interaction between the negatively charged siRNA and the positively charged G0–C14. However, the siRNA/G0-C14 complex might not be stable because the electrostatic interaction may be easily weakened by the high ionic strength condition of the body. Therefore, a hydrophobic polymer PLGA is used to cover the siRNA/G0-C14 complex via the hydrophobic interaction between the hydrophobic lipid tails of G0-C14 and the hydrophobic PLGA. It is worth noting that the cationic lipid G0-C14 plays a pivotal role in the fabrication of siRNA NPs. The cationic part of the G0-C14 should have enough positive charges to efficiently condense the negatively charged siRNA, and the lipid part of the G0-C14 should have enough hydrophobicity to interact with the hydrophobic PLGA. To reduce the nonspecific cellular uptake of the NPs and therefore prolong their blood circulation time in vivo, many neutral hydrophilic polymers have been used to modify the surfaces of the NPs66. Among them, PEG is the most popular polymer owing to its low toxicity and simple chemistry. Based on this, we introduced a PEG shell to cover the siRNA/G0-C14/PLGA core to increase the stability and blood circulation time of the siRNA NPs. The PEGylation process can be easily conducted by dropwise addition of the organic solution containing the siRNA/G0-C14/PLGA core to the aqueous solution containing the DSPE–PEG shell. For the introduction of targeting ligand such as S2P to the NPs, researchers just need to add the DSPE–PEG–S2P (or other targeting ligand-modified lipid) to the aqueous solution during the preparation of NPs. The targeting capability of the siRNA NPs is enabled by the S2P ligand; therefore, a high DSPE–PEG–S2P/DSPE–PEG ratio of 1:1 is recommended to maximize the targeting effect of the siRNA NPs to macrophages. Here, the DSPE–PEG–S2P/DSPE–PEG ratio of 1:1 was used for all in vitro and in vivo experiments. An optimal G0-C14/siRNA weight ratio was used to ensure the condensation of the siRNA by G0-C14; an optimal PLGA/siNRA weight ratio was used to ensure the protection of siRNA by PLGA; an optimal lipid–PEG/siRNA weight ratio was used to ensure the formation of stable siRNA NPs. In addition, an optimal stirring speed was used to achieve NPs with high siRNA EE and proper size; an optimal incubation time was used to ensure the formation of siRNA NPs and avoid the degradation of the siRNA.
To remove the small amount of organic solvent in the NP solutions, the NPs are further washed several times with water using an ultrafiltration purification method. We chose the ultrafiltration purification method on the basis of the following considerations. First, it saves a lot of time since the purification can normally be done in 1 or 2 h. Short operation time lowers the risk of degradation of siRNA during the purification process, and is especially important if the NP platform is used to deliver very unstable RNA therapeutics such as mRNA. Second, the concentration of the NPs can be easily adjusted. The ultrafiltration can result in a highly concentrated solution of NPs, which can be easily redispersed in any buffers to obtain NPs with any desired concentration according to different needs. This is quite useful when the NPs are applied in vivo, where high concentrations of NPs are normally required. In addition, our siRNA NP solution can be frozen and kept in the freezer for a few weeks, and then thawed for use. The thawed siRNA NPs show no substantial changes in activity, size and polydispersity index (PDI). We used a 20 mL glass vial to produce 400 µL of NPs (siRNA 5 µM). If a larger volume of NPs (e.g., 2 mL) is required, the synthesis can be scaled up to produce 2 mL of NPs by using five or more glass vials (20 mL).
Our polymer–lipid hybrid NPs can also be used as efficient delivery platforms for mRNA30,31. However, slight modifications are necessary for the preparation of mRNA NPs compared with the siRNA NP formulation presented here. The mRNA molecules are much larger than the siRNA molecules, and therefore the mRNA/G0-C14 weight ratio needs to be optimized to ensure the sufficient condensation of the mRNA by the cationic lipids. Compared with the preparation of siRNA NPs, where the siRNA/G0-C14 ratio of 1:10 is required, the preparation of mRNA NPs needs a larger amount of cationic G0-C14 lipids (mRNA/G0-C14 weight ratio,1:15). In addition, the stability of large and single-strand mRNA is much lower than that of small and double-strand siRNA. Therefore, the working area and all the tools need to be cleaned carefully with RNaseZAP to remove all the RNase contamination before starting to work with mRNA. Meanwhile, it is important to maintain the mRNA and mRNA NPs at a low temperature of 4 °C or below since mRNA is prone to degradation at room temperature.
DLS characterization of siRNA NPs (Steps 53–58)
It has been previously reported that particles with diameters greater than ~200 nm cannot pass the spleen’s fenestrations, which are typically <200–500 nm in width67,68. Thus, NPs with diameters in the range of 30–200 nm are ideal for systemic in vivo delivery because they can remain in circulation and avoid rapid renal clearance. To form siRNA NPs with ideal size, DLS is employed to monitor the size of each NP formulation. Most of the DLS instruments are able to detect NPs with sizes from 10 nm to 1 µm within a few minutes. Normally, 1.5 mL of sample is enough for detection, and it can be recycled for other tests. DLS can also be used to measure the zeta potential of the NPs. Any surface modifications of the NPs can lead to changes in zeta potential. Thus, by monitoring the changes in zeta potentials of the NPs, we can determine whether the surface modifications and alterations in surface charge are successful. If different users need to reproduce the NPs according to this protocol, DLS can be used to quickly confirm the successful production of the NPs.
TEM characterization of siRNA NPs (Steps 59–68)
Based on our design for the siRNA NPs, a core-shell structure is important with regard to protecting the siRNA from enzymatic degradation and prolonging the blood circulation time. TEM can clearly characterize the morphology and structure of the NPs. We thus use TEM to confirm the core-shell structure of the siRNA NPs. To improve the contrast of the sample in TEM images, 1% (wt/vol) of uranyl acetate is used to provide negative stain contrast in TEM images. The TEM images of the siRNA NPs in this protocol are produced using a JEOL 1200EX electron microscope at 80 kV.
Gel electrophoresis of siRNA NPs (Steps 69–91)
Our polymer–lipid hybrid NPs are designed to protect the loaded siRNA from enzymatic degradation. To confirm the protective effect of the NPs, the serum stability of siRNA NPs is assessed using agarose gel electrophoresis by monitoring the siRNA band. We incubate the siRNA-loaded NPs in a solution containing 50% (vol/vol) serum at 37 °C, a condition mimicking the in vivo environments, for different time periods. Theoretically, the enzymes in the serum can quickly degrade the siRNA at 37 °C upon direct exposure, resulting in the fading or disappearance of the siRNA band on agarose gel electrophoresis. To directly compare the siRNA that is loaded in the NPs with the free siRNA, the siRNA in the NPs is isolated before loading onto the agarose gel. To isolate the siRNA from the NPs, siRNA NPs are first pelleted using Amicon Ultra-15 centrifugal filter units (MWCO 100 kDa). Then, the pellet is dissolved in chloroform to destroy the core-shell structure of the NPs, releasing the incorporated siRNA, which is extracted from chloroform using 0.5 M NaCl containing 0.1% (wt/vol) sodium dodecyl sulfate6. The siRNA protection efficacy of siRNA by the polymer–lipid hybrid NPs is determined by gel electrophoresis using ethidium bromide-infused E-Gel 4% (wt/vol) agarose gels that are imaged under ultraviolet light.
Determination of siRNA EE (Steps 92-101)
Fluorescence detection is a highly sensitive technique widely used for the detection of compounds in solution even at very low concentrations. To determine the siRNA EE of the NPs, we label the siRNA with a fluorescent dye Dy647 (Dy647-siRNA). Alternatively, the siRNA EE of the NPs can be directly determined using ribogreen, one of the most sensitive fluorescent dyes for the detection of RNA in solution. Instead of ribogreen, we used dye-labeled siRNA (Dy647-siRNA) as this was also required for other experiments. The Dy647-siRNA is loaded into the NPs following the approaches we described above. The unloaded Dy647-siRNA can be easily removed in the ultrafiltration purification step. The encapsulation of fluorescent molecules into the cores of the NPs can result in changes in their fluorescence signals. Therefore, the Dy647-siRNA needs to be released from the NPs before it can be precisely determined by fluorescence detection. To this end, Dy647-siRNA-loaded NPs are dissolved in organic solvent dimethyl sulfoxide (DMSO), a solvent that can destroy the NPs by dissolving all the NP components, to release the encapsulated Dy647-siRNA. The fluorescence of the free siRNA can then be measured using a fluorescent plate reader, such as the Infinite M200 PRO spectrophotometer. The initial amount of Dy647-siRNA loaded into the NPs is also dissolved in DMSO, and its fluorescence is measured. The fluorescence of only DMSO is also measured and used as a blank control. All the measurements are conducted in triplicate. The siRNA EE of NPs can be calculated according to the following equation:
Evaluation of the stability of siRNA NPs (Steps 102–108)
The stability of the siRNA NPs in biological medium is critical to their long blood circulation, which allows the accumulation of siRNA NPs at lesional sites. To determine the stability of NPs in vitro, NPs are incubated with PBS containing 10% (vol/vol) serum (mimicking the in vivo environments) and their sizes are measured by DLS after different incubation time periods (up to 3 d). All the measurements are conducted in triplicate. The siRNA NPs after incubation for different time periods are considered to be stable at these conditions if their sizes show no notable changes compared with those of the freshly prepared NPs.
Characterization of endosomal escape of siRNA NPs (Steps 109–121)
The inefficient endosomal escape of siRNA NPs is reported to be a limiting factor for efficient siRNA delivery57,69. Therefore, the endosomal escape ability of NPs can increase siRNA delivery efficiency. To investigate the endosomal escape ability of our siRNA NPs, we first label NPs via labeling the encapsulated siRNA with a fluorescent dye Dy647 (red), so we can track the locations of siRNA NPs in the cells (we used HeLa cells) by monitoring the red fluorescence. Then, we use LysoTracker Green (green) to stain the late endosome whose locations in cells can be tracked via monitoring the green fluorescence. Upon incubation with Dy647-siRNA NPs for different time periods (1 or 4 h), the HeLa cells are first stained with LysoTracker Green for late endosome staining and then stained with Hoechst 33342 (blue) for nuclei staining. The cells are observed and imaged using an FV1000 laser scanning confocal microscope. The overlap of the red and green fluorescence signals of cells suggests that siRNA NPs are trapped inside the endosomes. The successful endosomal escape of siRNA NPs is defined by the separation of the red and green fluorescence signals of the cells.
In vitro evaluation of gene silencing by siRNA NPs (Steps 122–131)
Before the evaluation of siRNA NPs in vivo, we assess the gene silencing ability of siRNA NPs in vitro. To this end, one of the most widely used reporter gene systems based on luciferase-expressing HeLa (HeLa-Luc) cells is used for gene silencing assessment. Normally, the gene silencing efficiency of siRNA NPs can be determined by measuring the protein expression levels using western blot analysis, which is relatively costly and time consuming. The advantage of the HeLa-Luc system is that the gene silencing efficiency of the siRNA NPs can be quickly determined by measuring the bioluminescence using a plate reader or an In-Vivo Xtreme imaging system with a charge-coupled device (CCD) camera. A luciferase substrate must be added to the cells and incubated for 5 min before the measurements. Both quantitative data and images can be generated and used for gene silencing efficiency analysis. The gene silencing of our siRNA NPs is evaluated by incubating the siLuc NPs with HeLa-Luc cells for 24 h in 12-well plates using RPMI 1640 medium containing 10% (vol/vol) FBS as the culture medium. Upon 24 h incubation, the bioluminescence of the cells is measured using an In-Vivo Xtreme imaging system with a CCD camera. The bioluminescence signal of cells treated with PBS is measured and used as a PBS control. The treatment of PBS should not lead to any silencing effect of the Luc gene. All the measurements are conducted in triplicate. To quantify the gene silencing efficiency, the average radiance (photons per second per cm2 per steradian) within regions of interest is quantified using Bruker MI SE software.
In vitro viability, apoptosis and proliferation assessment of siRNA NPs (Steps 132–164)
Biocompatibility is a prerequisite in the development of NPs as safe in vivo siRNA delivery systems. In addition to the in vivo safety issues, the toxicity of the delivery systems can also interfere with the in vitro evaluations. For example, a false gene silencing effect can be achieved after incubating toxic siLuc NPs with HeLa-Luc cells. This can be ascribed to the fact that the toxicity of siLuc NPs can also decrease Luc expression by suppressing the growth of HeLa-Luc cells. To evaluate the safety of our siRNA NPs, we first investigate their in vitro toxicity. Different cells, including HeLa-Luc, RAW 264.7 and HEK-293 cells, are incubated with various amounts of siLuc NPs in 96-well plates for 24 h, after which the medium of the cells is replaced with a fresh medium and incubated for another 48 h. After the addition of the alamarBlue cell viability and proliferation reagent, the viability of the cells can be determined by measuring the absorbance at 570 nm using a plate reader. The absorbance of alamarBlue-treated medium is also measured and used as a blank control. The absorbance of cells treated with PBS is measured and used as a PBS control. The treatment of PBS should not lead to any cytotoxicity in the cells. All the measurements are conducted in five replicates. The viability of NP-treated cells can be calculated according to the following equation:
Some reagents can induce cell apoptosis, especially early apoptosis, which may not be detected by a cell viability reagent. We therefore also assess the apoptosis of cells treated with our siRNA NPs. RAW 264.7 cells are incubated with siLuc NPs containing 50 nM siLuc in 24-well plates for 24 h, after which the medium of the cells is replaced with a fresh medium and incubated for another 48 h. After the double staining with Annexin V (AV)-FITC and propidium iodide (PI) (Annexin V-FITC Apoptosis Detection kit), the apoptosis of the cells can be determined by flow cytometry. The apoptosis of cells treated with PBS is measured and used as a PBS control. The treatment of PBS should not lead to any pronounced apoptosis to the cells. All the measurements are conducted in triplicate. The early apoptosis of cells is defined by AV-FITC positive and PI negative, and the late apoptosis or dead cells are defined by both FITC AV and PI positive.
To investigate whether the siRNA NPs have an adverse effect on the growth of cells that is not detectable in the short term, we evaluate the proliferation of siRNA NPs-treated cells over 7 d. RAW 264.7 cells are incubated with siLuc NPs containing 50 nM siLuc in 96-well plates for 24 h, after which the medium of the cells is replaced with a fresh medium and incubated for another 7 d. After the addition of the alamarBlue cell viability and proliferation reagent, the proliferation of cells can be determined by measuring the absorbance at 570 nm using a plate reader. Notably, alamarBlue is a nontoxic agent that can be applied repeatedly on the same samples, allowing continuous monitoring of the samples70. Thus, the proliferation of cells is monitored every day, and the medium containing alamarBlue reagent is replaced by a fresh medium after each measurement. The absorbance of alamarBlue-treated medium is also measured and used as a blank control. The absorbance of cells treated with PBS is measured and used as a PBS control. The treatment of PBS should not lead to any inhibition to the growth of cells. All the measurements are conducted in five replicates. The proliferation of the NP-treated cells can be calculated according to the following equation:
In vivo evaluation of the therapeutic effect of siRNA NPs (Steps 165–189)
To assess the in vivo therapeutic effect of siRNA NPs for the treatment of atherosclerosis, we first establish a mouse model of atherosclerosis. It is well documented that the Ldlr gene-deficient mice can easily develop atherosclerotic plaques without any surgeries or injections by only feeding the mice with a high-fat Western diet (WD) for 6–8 weeks. Therefore, 8- to 10-week-old male mice with deficient Ldlr gene (Ldlr−/−) are obtained from Jackson Laboratory, and fed with WD for 8 weeks to develop the atherosclerotic plaques. It is worth noting that the influence of sex may need to be considered in many mouse models for human diseases71. We have used 8- to 10-week-old male mice with deficient Ldlr gene (Ldlr−/−) for the evaluation of the therapeutic effect of siRNA NPs. However, we speculate that this procedure would also be applicable to female mice since all the atherosclerosis-related phenotypes, including cholesterol, high-density lipoprotein, low-density lipoprotein, triglycerides and apolipoprotein B, elevate similarly in both male and female Ldlr knockout mice as demonstrated by Jackson Laboratory (supplier of laboratory mice). Therefore, sex should not have an important influence on the results generated by this protocol. The mice are randomly divided into three groups (with seven to nine mice per group) and injected with two doses of PBS, control siRNA NPs or siCamk2g NPs per week, respectively. After 4 weeks of treatment, the mice are euthanized with CO2 and their hearts with attached aortic roots are either fixed in 4% (wt/vol) paraformaldehyde followed by paraffin embedding or treated with optical cutting temperature compound and frozen. Sections of the aortic root with different sizes are isolated from the paraffin-embedded or frozen samples for the analysis of atherosclerotic lesions.
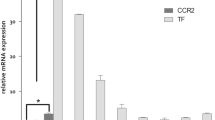
According to the proposed therapeutic mechanism of our siCamk2g NPs (Fig. 1), the treatment of siCamk2g NPs can knock down the expression of CaMKIIγ, resulting in the activation of the MerTK pathway in macrophages of the atherosclerotic lesions. To validate this mechanism, the expressions of CaMKIIγ and MerTK in lesional macrophages are characterized by immunofluorescence imaging. After blocking with bovine serum albumin (BSA), the obtained aortic root sections are first stained with primary anti-Mac2 and anti-MerTK and subsequently stained with fluorescently labeled secondary antibodies after PBS rinses. The nuclei of the macrophages are further stained with 4′,6-diamidino-2-phenylindole phenylindole (DAPI). The stained sections are imaged by a Zeiss epifluorescence microscope, and the images are analyzed by FIJI software. For statistical analysis, n = 7–9 mice per group.
Then, to further validate that the activation of the MerTK pathway can promote the efferocytosis of the lesional macrophages, the apoptotic cells in aortic root sections are detected by a TUNEL assay, a method for detecting apoptotic cells, using immunofluorescence imaging. The aortic root sections are first stained with TUNEL staining reagents and washed with PBS. Then, the sections are blocked with BSA, stained with primary anti-Mac2 antibody, incubated with fluorescently labeled secondary antibodies and stained with DAPI. The stained sections are imaged using a Zeiss epifluorescence microscope, and the images are analyzed using FIJI software. The apoptotic cells (TUNEL+) should be surrounded by Mac2+ macrophages when efferocytosis occurs. In contrast, isolated apoptotic cells have condensed nucleus and low antibody reactivity and are not associated with neighboring macrophages. Thus, the lesional efferocytosis can be quantified as the ratio of TUNEL+ cells (red) in contact with Mac2+ macrophages (green) to isolated TUNEL+ cells. For statistical analysis, n = 7–9 mice per group.
Finally, to confirm that the promoted efferocytosis can improve the plaque stability by reducing plaque necrosis and inhibiting fibrous cap thinning, the necrotic core area and fibrous cap thickness of the plaques in aortic root sections are determined by picrosirius red staining-based microscopy imaging. The picrosirius red staining is a widely used histological method to visualize collagen in paraffin-embedded tissue sections. Thus, the collagen of aortic root sections is stained with picrosirius red and imaged with light microscopy. The necrotic core area can be obtained directly from the images; the fibrous (collagen) cap thickness is quantified from one midpoint and two shoulder regions of the plaque and normalized to the lesion size72. For statistical analysis, n = 7–9 mice per group.
In vivo evaluation of the toxicity of the siRNA NPs (Steps 190–198)
The biosafety of the siRNA NPs is essential for their use in vivo. We therefore investigate the in vivo toxicity of the siRNA NPs under the same conditions (e.g., dose, frequency) as the therapeutic effect assay. Six-week-old female BALB/c mice obtained from Jackson Laboratory are injected intravenously with two doses of PBS, or S2P50 siCamk2g NPs (1 nmol siRNA per injection) per week for 4 weeks. The mice are euthanized by CO2 inhalation 3 d after the last administration. To evaluate whether the siRNA NPs can cause any damage to the organs of the mice, the major organs are fixed in 4% (wt/vol) paraformaldehyde followed by paraffin embedding. Sections with different sizes are isolated from the paraffin-embedded samples and subjected to H&E staining. In addition, the blood from the mice is collected and various hematological parameters are evaluated, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), hematocrit (HCT), hemoglobin (Hb), lymphocyte count (LY), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), neutrophil count (NPC), red blood cell count (RBC) and white blood cell count (WBC). For statistical analysis, n = 3 mice per group.
Recommended controls in the procedures
We recommend preparing the following control samples:
-
Step 71, untreated extracted siRNA control: extracted siRNA from siRNA S2P50 NPs without incubation with serum
-
Step 73, untreated siRNA control: free siRNA without incubation with serum (incubate with PBS instead)
-
Step 99, blank control: transfer 90 µL of 95% (vol/vol) DMSO solution to the 96-well plate
-
Step 107, untreated siRNA NPs control: the freshly prepared siLuc NPs without incubation with PBS containing 10% (vol/vol) serum
-
Step 124, PBS control (or 0 nM siLuc): for the bioluminescence signal of cells treated with PBS
-
Step 132, blank control: the absorbance of medium without any cells
-
Step 134, PBS control: the absorbance of cells treated with PBS
-
Step 145, PBS control: the apoptosis of cells treated with PBS
-
Step 151, blank control: unstained cells; AV-FITC control: cells stained with AV-FITC (no PI); PI control: cells stained with PI (no AV-FITC)
-
Step 153, blank control: the absorbance of medium without cells
-
Step 176, PBS-treated control: gene silencing of sections from mice treated by PBS; siCtrl NPs treated control: gene silencing of sections from mice treated by S2P50 siCtrl NPs
-
Step 185, PBS-treated control: efferocytosis of sections from mice treated by PBS; siCtrl NPs treated control: efferocytosis of sections from mice treated by S2P50 siCtrl NPs
-
Step 189, PBS-treated control: plaque stability of sections from mice treated by PBS; siCtrl NPs treated control: plaque stability of sections from mice treated by S2P50 siCtrl NPs
-
Step 198 PBS-treated control: H&E staining of sections from mice treated by PBS; hematological parameters of the blood from mice treated by PBS
Expertise needed to implement the protocol
This protocol describes all the procedures for each experiment in detail and is easy to follow. Any researchers trained in nanotechnology, gene delivery or biochemistry should be able to implement the protocol.
Limitations
The highly stable lipid–PEG shell of the NPs can reduce their cellular uptake, making them less suitable for in vitro or ex vivo applications, especially in situations where fast gene transfection is required. One solution to this issue is the introduction of targeting ligands. As demonstrated in this protocol, an S2P ligand is employed to improve the specific uptake of NPs by macrophages26. Another solution to this issue is the replacement of lipid–PEG. We have previously established that lipid–PEGs with long and saturated lipophilic tails had a long dissociation half-life (t1/2) when presented on NPs73. Therefore, the cellular uptake efficiency of the NPs can be tuned by using lipid–PEGs with short or unsaturated lipophilic tails. Despite the encouraging preclinical results that have been achieved using our polymer–lipid hybrid NP platform, the translation of our platform to clinical application needs further optimization. For example, the cationic lipids in our platform are biocompatible with most of the applications. However, in settings that require a high concentration of cationic lipids for NP synthesis, toxicity becomes an issue. Although negligible cytotoxicity was observed in cells treated with our NPs (siRNA 6.25–50 nM), we speculate that cytotoxicity >20% could be observed with siRNA doses >50 nM. This issue might be addressed by developing novel stimuli-responsive cationic lipids that can be degraded in the presence of various internal stimuli74.
Materials
Biological materials
-
Mouse model of interest
Caution
All experiments involving live animals must conform to the requirements of your institutional animal care and use committee (IACUC) and be performed strictly following protocols that are approved by IACUC. All our procedures were conducted in accordance with the protocols reviewed and approved by IACUC at Harvard Medical School and Columbia University Irving Medical Center. To produce the results shown here, we used:
-
Ldlr−/− mice, 8- to 10-week-old male mice (Jackson Laboratory, B6.129S7-Ldlrtm1Her/J, stock no. 002207)
-
BALB/c mice, 6-week-old female BALB/c mice (Jackson Laboratory, BALB/cJ, stock no. 000651)
-
-
Cells of interest
Caution
It is important to make sure that all the cell lines used in your research have no issues of cell line misidentification and cross-contamination. Our cell lines used in all the procedures had no issues of cell line misidentification and cross-contamination according to the latest report provided by the International Cell Line Authentication Committee (http://iclac.org/databases/cross-contaminations)
Caution
All the cell lines used in your research should be tested periodically to ensure they are authentic and not infected with mycoplasma. Our cell lines used in all the procedures were free of mycoplasma as determined by the Mycoplasma Detection Kit (Thermo Fisher Scientific, M7006). To produce the results shown here, we used:
-
HeLa-Luc cells (a HeLa cell line stably transfected with a firefly luciferase reporter gene, Sigma-Aldrich, cat. no. 11033106); RRID: CVCL_293975 (ECACC, cat. no. 11033106)
-
RAW 264.7 (ATCC, TIB-71); RRID: CVCL_0493 (ATCC, cat. no. TIB-71)
-
HEK-293 (ATCC, CRL-1573); RRID: CVCL_0045 (ATCC, cat. no. CRL-1573)
-
Reagents
-
G0 (amine) (Sigma-Aldrich, cat. no. 412368, CAS no. 155773-72-1)
-
MeOH (Sigma-Aldrich, cat. no. 322415, CAS no. 67-56-1)
Caution
MeOH is vaporizable and harmful to naked eyes. Work with personal protective equipment when handling MeOH (e.g., safety goggles and face shields).
-
1,2-epoxytetradecane (epoxide) (Sigma-Aldrich, cat. no. 260266, CAS no. 3234-28-4)
Caution
1,2-epoxytetradecane can cause severe damage to the respiratory system. Perform all operations under a chemical fume hood.
-
Dichloromethane (CH2Cl2; Sigma-Aldrich, cat. no. 34856, CAS no. 75-09-2)
Caution
Be careful when transferring CH2Cl2 involving cannulas and needles (syringe). Piercing the needles containing a residual amount of CH2Cl2 into tissues can cause severe injuries.
-
Ammonium hydroxide solution (NH4OH, Sigma-Aldrich, cat. no. 338818)
Caution
NH4OH can cause damage to the respiratory system. Handle this reagent under a chemical fume hood.
-
Silica gel (high-purity grade, average pore size 60 Å, 70–230 mesh, 63–200 μm, Sigma-Aldrich, cat. no. 60741-1KG)
-
Silica-gel-coated plates (Millipore Sigma, cat. no. MC1156840001)
-
DSPE–PEG–Mal (Nanocs, cat. no. PG2-DSML-3k, 3400 Da)
-
DSPE–PEG (Avanti Polar Lipids, cat. no. 880320, 3400 Da)
-
Targeting peptide CRTLTVRKC (S2P; GLS Biochem Systems, cat. no. 65235)
-
Nuclease-free water (HyPure water) (Fisher Scientific, cat. no. SH3053801)
-
RNaseZAP (Fisher Scientific, cat. no. AM9785)
-
Acetone (Sigma-Aldrich, cat. no. 650501, CAS no. 67-64-1)
Caution
Flammable and vaporizable acetone is harmful to the central nervous system. Work with acetone under a fume hood.
-
N,N-Dimethylformamide (Sigma-Aldrich, cat. no. 227056, CAS no. 68-12-2)
Caution
N,N-Dimethylformamide may cause abdominal pain, headache, skin problems, constipation, nausea and vomiting. Wear personal protective equipment (e.g., eyeshields, face shields, laboratory coats and gloves) during operation.
-
50:50 Poly(DL-lactide-co-glycolide) (PLGA; LACTEL Absorbable Polymers, cat. no. B6013-2, Inherent Viscosity dL/g: 0.55–0.75)
-
1× PBS (Fisher Scientific, cat. no. SH3025601, pH 7.4)
-
NaOH solution, 1 M (Sigma-Aldrich, cat. no. 1091371000)
-
HCl solution, 1 M (Sigma-Aldrich, cat. no. 1506961000)
-
UAR-EMS uranyl acetate (Electron Microscopy Sciences, cat. no. 22405)
-
FBS (Thermo Fisher Scientific, cat. no. 26140095)
-
Chloroform (Sigma-Aldrich, cat. no. C2432, CAS no. 67-66-3)
Caution
Chloroform is toxic and can cause severe health problems, including central nervous system damage, irritation of skin and eyes, etc. Perform all operations under a chemical fume hood.
-
Ethidium-bromide-infused E-Gel 4% (wt/vol) agarose gel (Thermo Fisher Scientific, cat. no. G501804)
-
10× BlueJuice Gel Loading Buffer (Thermo Fisher Scientific, cat. no. 10816015)
-
DMSO (Sigma-Aldrich, cat. no. 276855, CAS no. 67-68-5).
-
RPMI 1640 medium (Thermo Fisher Scientific, cat. no. 11875119)
-
Penicillin–streptomycin (10,000 U/mL penicillin, 10,000 μg/mL streptomycin; Thermo Fisher Scientific, cat. no. 15140122)
-
Hoechst 33342 (Thermo Fisher Scientific, cat. no. H1399)
-
LysoTracker Green (Thermo Fisher Scientific, cat. no. L7526)
-
Steady-Glo Luciferase reagent (Promega Corporation, cat. no. E2520)
-
Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific, cat. no. 11965092)
-
AlamarBlue cell viability reagent (Thermo Fisher Scientific, cat. no. DAL1100)
-
Annexin V-FITC Apoptosis Detection kit (Thermo Fisher Scientific, cat. no. V13245)
-
‘Western’ purified atherogenic diet (Envigo, TD.88137 Adjusted calories diet, 42% kcal from fat)
-
Paraformaldehyde solution, 4% (wt/vol) in PBS (Fisher Scientific, cat. no. AAJ19943K2)
-
BSA (Sigma-Aldrich, cat. no. A2153, CAS no. 9048-46-8)
-
10× Tris-buffered saline (Bio-Rad Laboratories, cat. no. 1706435)
-
Tween 20 (Sigma-Aldrich, cat. no. P2287)
-
Goat serum (Sigma-Aldrich, cat. no. G9023)
-
Triton X-100 (Sigma-Aldrich, cat. no. X100)
-
DAPI (Sigma-Aldrich, cat. no. D8417, CAS no. 28718-90-3)
-
Picrosirius red (Polysciences, cat. no. 24901A)
-
Anti-luciferase RNA (siLuc), Dy647-labeled siLuc (Dy647-siLuc) and anti-CaMKIIγ RNA (siCamk2g) (Dharmacon, custom siRNA, siRNA sequence can be found in Table 1)
Table 1 The siRNA sequences used in this protocol
Equipment
-
Heating plate (Isotemp Hot Plate, Fisher Scientific, cat. no. HP88857296)
Caution
Avoid contacting heated part of the plate during heating operation, which can cause severe burns.
-
25 mL round-bottom flask (Chemglass, cat. no. CG-618-01)
-
500 mL round-bottom flask (Chemglass, cat. no. CG-618-09)
-
Blood collection tube (Greiner Bio-One, cat. no. 454012)
-
Amicon Ultra-15 centrifugal filter units (Sigma-Aldrich, cat. no. UFC9003, MWCO 3 kDa)
-
10 mL glass vial (Chemglass, cat. no. CG-4908-05).
-
20 mL glass vial (Chemglass, cat. no. CG-4908-01)
-
Gel loading tips (Fisher Scientific, cat. no. 02-707-139)
-
Carbon support copper grids (Electron Microscopy Sciences, cat. no. FCF200-CU)
-
96-well plate (Fisher Scientific, cat. no. CLS3595)
-
Nunc glass-bottom dish (Thermo Fisher Scientific, cat. no. 150680)
-
12-well plate (Fisher Scientific, cat. no. CLS3512)
-
Six-well plate (Fisher Scientific, cat. no. CLS3516)
-
Syringes (BD, 1/2 cc insulin syringe U-100, cat. no. BD 329424)
-
Oil bath (Chemglass, cat. no. CG-8276-190)
-
Rotary evaporator (Buchi Labortechnik AG, cat. no. R-124)
-
Centrifuge (Thermo Scientific Sorvall Legend X1R Centrifuge, cat. no. 75004263)
-
Orion Star A221 Portable pH Meter (Sigma-Aldrich, cat. no. STARA221)
-
Nanodrop spectrophotometer (Thermo Fisher Scientific, NanoDrop 1000)
-
Sonication bath (Fisher Scientific, cat. no. FB11207)
-
Digital vortexer (Fisher Scientific, cat. no. 0215370)
-
DLS measurement (Brookhaven Instruments Corporation, ZetaPALS)
-
Transmission electron microscopes (JEOL, 1200EX)
-
E-Gel Power Snap Electrophoresis Device (Thermo Fisher Scientific, cat. no. G8100)
-
Confocal laser scanning microscope (Olympus, FLUOVIEW FV1000)
-
Flow cytometer (BD, LSR Fortessa)
-
Bioluminescence imaging system (Bruker, In-Vivo Extreme II Optical/X-ray system)
-
Plate reader (Tecan, Infinite M200 PRO)
-
GraphPad (https://www.graphpad.com)
-
FIJI software (https://imagej.net/Fiji)
Reagent setup
HEPES solution (Thermo Fisher Scientific, 1 M, pH 7.0–7.6, cat. no. 15630080)
To adjust the pH of HEPES solution to pH 6.5, introduce HCl while monitoring the pH with a pH meter. If the pH goes too low, raise it by carefully adding NaOH.
Caution
Users should exercise extreme caution when working with NaOH and HCl. Wear gloves, eye protection, face shields and laboratory coats during operation. The buffer can be stored at 4 °C for up to 6 months.
RPMI 1640 cell culture medium
To prepare the cell culture medium, add 50 mL FBS and 5 mL penicillin–streptomycin into 450 mL RPMI 1640 medium. The as-prepared cell culture medium contains 10% (vol/vol) FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. The medium can be stored at 4 °C for up to 6 weeks.
1× Tris-buffered saline with 0.1% (vol/vol) Tween 20
Dilute 100 mL 10× Tris-buffered saline with 900 mL deionized water, and add 1 mL Tween 20. A brief sonification will help to homogeneously mix the as-prepared 1× Tris-buffered saline with 0.1% (vol/vol) Tween 20. The buffer can be stored at 4 °C for up to 3 months.
Blocking buffer (1× PBS containing 5% (vol/vol) normal serum and 0.3% (vol/vol) Triton X-100)
To prepare a 10 mL blocking buffer, add 0.5 mL normal goat serum into 9.5 mL 1× PBS. While stirring, add 30 µl Triton X-100 into the above solution. The buffer should be prepared fresh before use.
Antibody dilution buffer (1× PBS containing 1% (wt/vol) BSA and 0.3% (vol/vol) Triton X-100)
To prepare a 10 mL antibody dilution buffer, add 30 µl Triton X-100 into 10 mL 1× PBS. Then add 0.1 g BSA to the solution. Make sure the solution is homogeneously mixed. The buffer should be prepared fresh before use.
Anti-CaMKIIγ (1:400, Novus Biologicals, cat. no. NBP2-15685); RRID: AB_2892990 (Novus, cat. no. NBP2-15685)
Dilute the primary antibody with antibody dilution buffer. The buffer should be prepared fresh before use.
Anti-Mac2 antibody (1:10, 000, Cedarlane Labs, cat. no. CL8942AP); RRID: AB_2814900 (Fluidigm cat. no. 3153026B)
Dilute the primary antibody with antibody dilution buffer. The buffer should be prepared fresh before use.
Anti-MerTK (1:500, R&D Systems, cat. no. AF591); RRID: AB_2098565 (R and D Systems, cat. no. AF591)
Dilute the primary antibody with antibody dilution buffer. The buffer should be prepared fresh before use.
Alexa Fluor 647 Goat–anti Mouse IgG (Life Technologies, A-28181); RRID: AB_2536165 (Thermo Fisher Scientific, cat. no. A28181)
Dilute the second antibody with antibody dilution buffer. The buffer should be prepared fresh before use.
Mouse model of atherosclerosis
To establish a mouse model of atherosclerosis, purchase 8- to 10-week-old male mice with deficient Ldlr gene (Ldlr−/−) from Jackson Laboratory. Then, feed the mice with a high-fat WD for 8 weeks to develop atherosclerotic plaques26.
Procedure
Synthesis of cationic lipid (G0-C14)
Timing ~3 d
Critical
This procedure should result in ~1.9 g of purified cationic lipid.
-
1
Weigh 1.0 g (1.936 mmol, 1 eq.) of G0 (amine), and add it to a 25 mL round-bottom flask.
Critical step
The G0 is 20% (wt/vol) in MeOH, so 1.0 g of G0 is equal to 1.0/0.2 = 5.0 g G0 solution.
-
2
Weigh 2.878 g (13.552 mmol, 7 eq.) of 1,2-epoxytetradecane (epoxide), and add it to the round-bottom flask containing G0.
Critical step
The 2-epoxytetradecane has a purity of 85% (wt/wt), so 2.878 g of 1,2-epoxytetradecane is equal to 2.878/0.85 = 3.386 g crude 1,2-epoxytetradecane. The ratio of the amines (G0) and hydrocarbon tails (C14) is critical for the construction of our platform. G0 has four primary amines that can maximally react with eight lipid tails (1,2-epoxytetradecane). The feeding ratio of epoxide and G0 is 7:1 here because we want to form products with seven lipid tails as these materials display high siRNA delivery efficiency.
-
3
Add a magnetic bar to the round-bottom flask, and equip the flask with a drying tube.
-
4
Place the flask in an oil bath supported by a hot-plate magnetic stirrer, and start stirring the reaction solution.
-
5
Set the heating temperature to 90 °C, and start heating the solution.
-
6
After 30 min, when all of the solvent (MeOH) evaporates, replace the drying tube with an argon balloon.
Critical step
Argon protection is mandatory due to the involvement of amine in the reaction.
-
7
Stir the reaction solution at 90 °C for 48 h under argon atmosphere.
Critical step
The viscosity of the reaction mixture increases with the evaporation of the MeOH. Ensure your magnetic stir bar is large enough so that it continues stirring in the increased-viscosity solution.
-
8
After the completion of the reaction, remove the balloon and the magnetic stir bar.
Critical step
This is a solvent-free reaction, so there is no need to remove solvent after the reaction.
-
9
Add 2.0 mL of CH2Cl2 to dissolve the reaction mixture.
-
10
Purify the product by flash silica gel column chromatography (silica gel, high-purity grade). Load the crude product solution to the silica column (diameter 40 mm, height 400 mm), remove the unreacted starting materials using CH2Cl2 and then elute the product using 75:22:3 CH2Cl2/MeOH/NH4OH. The purified G0-C14 contains seven lipid tails as calculated from the 1H NMR spectrum60.
-
11
Collect the eluents in 20 mL fractions using 30 mL tubes; around 25 fractions should be collected. Monitor the fractions by thin-layer chromatography using silica-gel-coated plates and 75:22:3 CH2Cl2/MeOH/NH4OH as eluent (Rf is ~0.3 for product). Visualize the eluted product on the thin-layer chromatography plate by I2.
-
12
Product is expected to appear between fraction No. 5 and No. 10. Combine and transfer all the fractions containing pure product to a clean 500 mL round-bottom flask. Remove all the solvents using a rotary evaporator at 150 mbar and 25 °C in a water bath. The maximum temperature can be used is 45 °C, and it normally takes 1–2 h to get the oily product.
-
13
Place the flask under a high vacuum for 24 h to remove any residual solvent.
-
14
Weigh the pure G0-C14, calculate the yield and store it at −20 °C.
Pause point
The pure G0-C14 can be stored at −20 °C for at least 1 year.
Synthesis of ligand-modified lipid–PEG (DSPE–PEG–S2P)
Timing ~3 d
Critical
This procedure should result in ~105 mg of purified ligand-modified lipid.
-
15
Weigh 200.0 mg (0.059 mmol, 1.0 eq.) of DSPE–PEG–Mal, and add it to a 10 mL round-bottom flask.
-
16
Weigh 63.7 mg (0.059 mmol, 1.0 eq.) of targeting peptide CRTLTVRKC (S2P), and add it to the round-bottom flask containing DSPE–PEG–Mal.
-
17
Add 2 mL of 1× HEPES buffer (pH 6.5) to dissolve the starting materials.
Critical step
Controlling the pH by HEPES buffer is important for the dissolution of S2P peptide and maximal reaction efficiency.
-
18
Add a magnetic bar to the round-bottom flask, and equip the flask with an argon balloon.
-
19
Place the flask on a hot-plate magnetic stirrer, and start stirring the reaction solution.
-
20
Stir the reaction solution at room temperature (20–22 °C) for 48 h under argon atmosphere.
-
21
After the completion of the reaction, remove the balloon and the magnetic stir bar.
-
22
Pour the reaction solution into 30 mL of cold MeOH in a 50 mL Falcon tube to precipitate the product. Collet the precipitates by centrifugation (2,305g, 5 min, 4 °C), and wash the precipitate two times with 15 mL of cold MeOH.
Critical step
Alternatively, purify the product with ultrafiltration using Amicon Ultra-15 centrifugal filter units (MWCO 3 kDa). After five rounds of washes, collect the filtrates containing pure product and remove the water by lyophilization.
-
23
Collect and transfer the precipitates to 2 mL Eppendorf tubes. Place the tubes under high vacuum for 24 h to remove any residual solvent.
-
24
Weigh the pure DSPE–PEG–S2P, calculate the yield and store it at −20 °C.
Pause point
The pure DSPE–PEG–S2P can be stored at −20 °C for at least 1 year.
Preparation of siRNA stock solution
Timing ~1 h
-
25
Order anti-luciferase RNA (siLuc), Dy647-labeled siLuc (Dy647-siLuc) and anti-CaMKIIγ RNA (siCamk2g) (Table 1). We recommend ordering 1.0 µmol of each siRNA to perform the following in vitro and in vivo procedures.
-
26
Upon receiving the siRNA, spin down the lyophilized siRNA product.
Critical step
Normally, the lyophilized siRNAs are light and have very small volumes; thus, they can be easily flushed out from the tubes by airflow when they are near the upper parts of the tubes. So, do not open the tubes before spin-down.
-
27
To make a siRNA stock solution of 100 µM, add 0.75 mL of nuclease-free water (HyPure water) to a tube containing 75 nmol siRNA.
-
28
Place the tube on a vortex for 30 s to dissolve the siRNA and spin down the solution. Repeat this process three times.
Critical step
It takes time to dissolve the siRNA in water, so make sure the siRNA is completely dissolved before it is used for further experiment. Otherwise, the concentration of siRNA cannot be precisely determined.
Critical step
Do not use a sonicator to facilitate the dissolution of the siRNA, because it can destroy the siRNA.
-
29
Transfer 1 µL of the obtained siRNA solution to a Nanodrop spectrophotometer to determine the concentration of the siRNA solution.
-
30
Aliquot the siRNA solution into 50 µL fractions, and store them at −20 °C.
Critical step
It is important to aliquot the siRNA solution to minimize repeated freeze–thaw cycles, which can degrade the siRNA. Notably, we have not observed notable degradation of siRNA with three freeze–thaw cycles.
Pause point
The siRNA aliquots can be stored at −20 °C for 1 year.
Preparation of siRNA-loaded NPs
Timing ~4 h
Critical
This procedure should produce ~400 µL of purified siRNA-loaded NPs (5 µM siRNA content). This scale is sufficient for most of the downstream characterization and in vitro and in vivo experiments. To perform downstream gel electrophoresis studies (Steps 69–91), this procedure will need to be scaled sevenfold. For serum stability studies (Steps 102–108), this procedure will need to be scaled twofold. For in vivo studies (Steps 165–170), this procedure will need to be scaled fivefold. See ‘Experimental design’ for advice on scaling the reaction.
-
31
Clean the working area and all the tools with RNaseZAP to remove all the RNase contamination.
Critical step
This is especially important if the NPs are used to deliver more unstable nucleic acid cargoes, such as mRNA.
-
32
Weigh 10 mg of G0-C14 (from Step 14), and add it to a 10 mL glass vial.
-
33
To make an acetone solution containing 2.5 mg/mL G0-C14, add 4 mL of acetone to a glass vial and place the vial in a sonication bath for 10 min to dissolve the G0-C14.
Critical step
The dissolution of G0-C14 in acetone is very slow even under sonication, so be patient to make sure that a clear solution is obtained (this usually takes 10 min). It is very difficult or impossible to dissolve the G0-C14 when its concentration is >2.5 mg/mL, so the maximum G0-C14 concentration is 2.5 mg/mL. In another study of our group, we have shown that acetone can be replaced by dimethylformamide31.
-
34
Weigh 20 mg of PLGA, and add it to a 10 mL glass vial.
-
35
To make an acetone solution containing 5 mg/mL PLGA, add 4 mL of acetone to the glass vial and place the vial in a sonication bath for 3 min to dissolve the PLGA.
-
36
Add a magnetic bar to a 20 mL glass vial, and rinse the vial with 10 mL of HyPure water; repeat the rinse three times.
Critical step
This wash step can reduce the RNase contamination of the vials and the magnetic bar.
-
37
Weigh 1 mg of hybrid lipid–PEGs containing different ratios of DSPE–PEG–S2P (from Step 24)/DSPE–PEG, and add it to the 20 mL glass vial. Select the composition for your intended application using Table 2. We recommend 1:1 ratio for atherosclerotic lesional macrophages-targeting application. The targeting capability of the siRNA NPs is enabled by the S2P ligand; therefore, a high DSPE–PEG–S2P/DSPE–PEG ratio of 1:1 is recommended to maximize the targeting effect of the siRNA NPs to macrophages. Here, the DSPE–PEG–S2P/DSPE–PEG ratio of 1:1 was used for all in vitro and in vivo experiments.
Table 2 The composition of different siRNA NPs -
38
To make an aqueous solution containing 0.1 mg/mL lipid–PEGs, add 10 mL of HyPure water to the glass vial and place the vial on a vortex for 30 s to dissolve the lipid–PEGs.
-
39
Place the glass vial on a magnetic stirrer, and start stirring the lipid–PEG solution at a stirring speed of 600 rpm.
-
40
Thaw the siRNA aliquots on the ice. One aliquot is required for the preparation of 400 µL of siRNA-loaded NPs (5 µM siRNA content).
Critical step
Thawing siRNA aliquots on ice can minimize the degradation of siRNA. Do not thaw the siRNA aliquots in a 37 °C water bath.
-
41
Transfer 20 µL of 100 µM stock siRNA solution (2 nmol) to a 1.7 mL Eppendorf tube.
Critical step
To prepare NPs containing different types of siRNA, just use different siRNA sequences (e.g., siLuc, Dy647-siLuc, siCamk2g) during the preparation of NPs. No further modifications of the procedures are required.
-
42
Add 100 µL of 2.5 mg/mL G0-C14 acetone solution (250 µg, from Step 33) to the tube to complex the siRNA. Mix by gentle pipetting (10 s) to form siRNA/G0-C14 complexes via the electrostatic interactions.
-
43
Add 500 µL of 5 mg/mL PLGA acetone solution (2.5 mg, from Step 35) to the tube. Mix by gentle pipetting (10 s) to achieve a homogeneous siRNA/G0-C14/PLGA solution.
-
44
Immediately add the siRNA/G0-C14/PLGA solution dropwise to the 20 mL vial containing 10 mL of the lipid–PEG solution (from Step 37) under stirring (600 rpm).
Critical step
The acetone solution should be added to the aqueous solution dropwise to allow the assembly of the core-shell structure of the polymer–lipid hybrid NPs. Use the gel loading tips to add the solution, because the speed of adding solutions can be more precisely controlled by this type of tips compared with the normal tips. In addition, immerse the tips in the aqueous solution during the addition instead of on the top of it.
-
45
After the completion of the addition, raise the stirring speed to 1,000 rpm and stir the solution at room temperature for 1 h.
Critical step
The stirring speed is important because vigorous stirring can facilitate the formation of the polymer–lipid hybrid NPs. In another study of our group, we have shown that the stirring time can be shortened to 30 min to minimize the degradation of the nucleic acid cargos at room temperature30.
-
46
Rinse the Amicon Ultra-15 centrifugal filter unit (MWCO 100 kDa) with 10 mL of HyPure water; repeat this rinse process three times.
-
47
After the completion of the assembly of siRNA NPs, transfer the NP solution (from Step 45) to the Amicon tube.
-
48
Concentrate the NP solutions by centrifugation for 20 min at 3,011g, 4 °C.
-
49
Wash the NPs by adding 10 mL of HyPure water to the Amicon tube; redisperse the NPs by pipetting. Concentrate the NP solutions by centrifugation for 20 min at 3,011g, 4 °C.
-
50
Repeat the wash step three times.
Critical step
The centrifugation requires a longer time after the first wash, so increase the time for centrifugation accordingly to make sure that most of the solution is removed by filtration at each wash.
-
51
After the final wash, add 400 µL of PBS (pH 7.4) to redisperse the siRNA NPs. This should result in an siRNA concentration of ~5 µM (RNA EE is measured in Steps 92–101).
Critical step
Control the final concentration of siRNA NP by adding a different volume of PBS according to different needs. For example, 400 µL of siRNA NPs with a high siRNA concentration (20 µM of siRNA) is required for Step 92. To this end, add 100 µL of PBS (pH 7.4) to redisperse the siRNA NPs to obtain 100 µL of siRNA NPs with ~20 µM siRNA; combine four batches of this solution to obtain 400 µL of siRNA NPs (20 µM of siRNA content).
-
52
Transfer the siRNA NPs (siRNA concentration, 5 µM, 400 µL) to a 1.7 mL Eppendorf tube, and put it on ice immediately.
Pause point
The siRNA NPs with siRNA concentration of 5 µM or higher can be used prepared fresh or stored at −20 °C for at least 1 month. One freeze–thaw cycle has no obvious influence on the effectiveness of the siRNA NPs; however, repeated freeze–thaw cycles should be avoided. If mRNA NPs are prepared, they can be used prepared fresh or stored at −80 °C for 2 weeks.
DLS characterization of siRNA NPs
Timing ~5 h
-
53
Prepare 400 µL of each siLuc NPs (S2P0, and S2P50 NPs, siLuc 5 µM) according to the procedures for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2).
-
54
Add 150 µL of S2P0, or S2P50 NPs (siLuc, 5 µM) to a 1.7 mL Eppendorf tube.
-
55
Add 1.35 mL of PBS (pH 7.4) to the tube to dilute the siLuc NPs.
-
56
Vortex and spin down the solution to achieve 1.5 mL of siLuc NPs (siLuc, 0.5 µM).
Critical step
The volume of the siLuc NPs has to be enough for the DLS measurement; otherwise, unreliable results could be obtained.
-
57
Transfer the siLuc NPs to a cuvette, and measure the size (intensity and distribution) of the siLuc NPs by DLS.
-
58
After the size measurement, recover the sample and measure its zeta potential.
TEM characterization of siRNA NPs
Timing ~6 h
-
59
Prepare 400 µL of each siLuc NPs (S2P0, S2P10, S2P30 and S2P50 NPs; siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2).
-
60
Put a filter paper on the bench.
-
61
Mount the carbon-coated copper grids in a tweezer, and rest the tweezer on the filter paper.
-
62
Place 20 µL of S2P0, S2P10, S2P30 or S2P50 NPs on the grid, and incubate at room temperature for 10 min.
Critical step
The volume can be adjusted in the range of 3–20 µL according to the concentration of the NPs.
-
63
After sample incubation, blot the sample with filter paper.
-
64
Wash the sample by placing 3 µL of ultrapure water on the grid.
Critical step
A second wash can be applied if too much salt is observed on TEM imaging.
-
65
Blot the water with filter paper.
-
66
Immediately place 3 µL of stain solution (UAR-EMS uranyl acetate replacement stain) on the grid for 10 s, and blot the stain solution with filter paper.
-
67
Dry the sample under air for >1 h.
-
68
When the sample is completely dry, image the sample using a JEOL 1200EX electron microscope at 80 kV.
Gel electrophoresis of siRNA NPs
Timing ~16 h
-
69
Prepare 2.8 mL of siLuc NPs (siLuc S2P50 NPs; siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2; this is a sevenfold-larger scale than reported in the above steps).
-
70
Prepare five Eppendorf tubes (1.7 mL); add 0.5 mL of FBS to each tube.
-
71
Add 0.5 mL of siLuc S2P50 NPs (siLuc 5 µM) to each of the five tubes containing serum. Prepare an untreated extracted siRNA control: extracted siRNA from siRNA S2P50 NPs without incubation with serum.
-
72
Mix the solution by vortexing, and spin down the solution.
Critical step
Use a solution containing as high as 50% (vol/vol) serum to mimic the in vivo environments.
-
73
Similarly, prepare five Eppendorf tubes (1.7 mL); add 0.5 mL of FBS to each tube. Add 0.5 mL of free siLuc PBS solution (siLuc 5 µM) to each of the five tubes containing serum. Mix the solution by vortexing, and spin down the solution. Prepare an untreated siRNA control: free siRNA without incubation with serum (incubate with PBS instead).
-
74
Incubate all five of the siLuc S2P50 NP samples and five free siLuc samples at 37 °C in a water bath.
-
75
After incubation for different time periods (0, 3, 6, 9 or 12 h), take out one siLuc S2P50 NP sample and one siLuc sample at each timepoint.
-
76
Transfer the NP samples to Amicon Ultra-15 centrifugal filter units (MWCO 100 kDa); concentrate the samples by centrifugation at 3,011g, 4 °C for 15 min.
Critical step
Adjust the time for centrifugation accordingly if necessary.
-
77
Transfer the concentrated NP samples to 1.7 mL Eppendorf tubes.
-
78
Add 0.5 mL of chloroform to each tube to dissolve the concentrated NP samples.
Critical step
It is critical to keep the volume of water in NP samples as low as possible, so the chloroform can efficiently destroy the core-shell structure of the NPs and release the encapsulated siRNA.
-
79
Extract the siLuc by adding 0.5 M NaCl containing 0.1% (wt/vol) sodium dodecyl sulfate to each tube containing chloroform-dissolved NP samples.
-
80
Vortex and spin down the solutions.
-
81
Collect the upper aqueous layers.
-
82
Repeat the extraction process three times.
-
83
Combine and transfer all the aqueous layers to 2 mL Eppendorf tubes.
-
84
Transfer the free siLuc samples to 2 mL Eppendorf tubes, and dilute them to the same volume as the NP samples.
-
85
Store all the five NP samples and five free siLuc samples at −20 °C.
Pause point
The samples can be stored at −20 °C for at least 2 weeks.
-
86
Unpack the ethidium-bromide-infused E-Gel 4% (wt/vol) agarose gel, and load the gel into a horizontal electrophoresis apparatus.
-
87
Add 20 µL of each of the ten siRNA samples to ten Eppendorf tubes (0.6 mL) along with control samples.
-
88
Add 2 µL of loading buffer to each tube; mix them by vortexing, and spin down.
-
89
Load 20 µL of each of the ten siRNA samples to the wells of the agarose gel.
-
90
Run the gel electrophoresis for 30 min using E-Gel Power Snap System (voltage 48 V).
-
91
After the completion of gel electrophoresis, visualize the agarose gel in an ethidium channel using a Syngene PXi imager.
Determination of siRNA EE
Timing ~4 h
-
92
Prepare 400 µL of Dy647-siLuc NPs (S2P0, and S2P50 NPs; Dy647-siLuc 20 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2).
Critical step
Conduct all the handling with Dy647-siLuc in the dark since the Dy647 is very sensitive to light exposure.
-
93
Add 5 µL of Dy647-siLuc S2P0 or S2P50 NPs to a 0.6 mL Eppendorf tube.
-
94
Add 95 µL of DMSO to the tube to dissolve the NPs and release the encapsulated Dy647-siLuc.
-
95
Mix the solution by vortexing, and spin down the solution (final feeding Dy647-siLuc, 1 µM).
Critical step
It is critical to completely dissolve the NPs in DMSO because the incomplete dissolution of the NPs can lead to a decrease in siRNA EE. Repeat the vortexing process if necessary.
-
96
Similarly, prepare a free Dy647-siLuc solution (Dy647-siLuc, 20 µM). Add 5 µL of free Dy647-siLuc to a 0.6 mL Eppendorf tube containing 95 µL of DMSO. Mix the solution by vortexing, and spin down the solution (final free Dy647-siLuc, 1 µM).
-
97
Prepare all the samples in six replicates.
-
98
Transfer 90 µL of each sample to a 96-well plate.
-
99
Prepare a blank control: transfer 90 µL of 95% (vol/vol) DMSO solution to the 96-well plate.
-
100
Measure the fluorescence intensities of all the samples using an Infinite M200 PRO spectrophotometer (excitation 645 nm, emission 680 nm).
-
101
The siRNA EE of NPs can be calculated according to the following equation:
$$\% \,{{{\mathrm{EE}}}} = 100 \times \left( {{{{\mathrm{sample}}}} - {{{\mathrm{blank}}}}} \right)/\left( {{{{\mathrm{load}}}} - {{{\mathrm{blank}}}}} \right).$$
Evaluation of the stability of siRNA NPs
Timing ~3 d
-
102
Prepare 800 µL of siLuc NPs (S2P0, and S2P50 NPs; siLuc, 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2. The procedure will need to be doubled to produce the required amount of NPs for this assay).
-
103
Add 200 µL of siLuc S2P0 or S2P50 NPs to a 2 mL Eppendorf tube. Prepare all the samples in triplicate.
-
104
Add 1.6 mL of PBS and 200 µL of FBS to each of the tubes to dilute the siLuc NPs.
-
105
Mix the solution by vortexing, and spin down the solution.
-
106
Incubate the siLuc NP solutions at 37 °C over 3 d. At different timepoints (0, 12, 24, 36, 48, 60 and 72 h), take out the siLuc NPs and transfer 1.5 mL of them to a cuvette.
-
107
Prepare an untreated siRNA NP control in triplicate: the freshly prepared siLuc NPs without incubation with PBS containing 10% (vol/vol) serum.
-
108
Monitor the sizes of the NPs by DLS, recover the samples and then continue incubating them at 37 °C.
Critical step
It is normal that the NPs in FBS show an extra 20 nm peak compared with the NPs in PBS, because this peak belongs to serum proteins in FBS.
Characterization of the endosomal escape of siRNA NPs
Timing ~30 h
-
109
Seed 1 mL of HeLa-Luc cells in Nunc glass-bottom dishes at a density of 2 × 105 cells per well (complete RPMI 1640 medium). Incubate the cells at 37 °C in a humidified incubator containing 5% (vol/vol) CO2 for 24 h.
-
110
Prepare 400 µL of Dy647-siLuc NPs (S2P50 NPs; Dy647-siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2)
-
111
Add 10 µL of Dy647-siLuc NPs to a 1.7 mL Eppendorf tube. Prepare the samples in duplicates.
-
112
Add 1 mL of RPMI 1640 medium to each of the tubes to dilute the NPs.
-
113
Mix the solution by pipetting gently.
-
114
Remove the cell medium (from Step 109), and add 1 mL of medium containing Dy647-siLuc NPs to each of the two duplicate wells, respectively. The final concentration of Dy647-siLuc in each well is 50 nM.
-
115
Put the cells back in the incubator.
-
116
Remove the NPs after 1 h or 4 h incubation. Wash the cells three times with PBS.
-
117
Stain the late endosome of the cells by adding 1 mL of medium containing 50 µM Hoechst 33342 (stock solution 1 mM), and incubate for 30 min.
Critical step
The incubation time can be varied from 30 min to 2 h depending on the cell type and density.
-
118
Remove the staining solution, and rinse the cells three times with PBS.
-
119
Stain the nuclei of the cells by adding 1 mL of medium containing 5 µg/mL Hoechst 33342 (stock solution 10 mg/mL), and incubate for 20 min.
Critical step
The nuclei can be easily over-stained, so decrease the incubation time if necessary.
-
120
Remove the staining solution, and rinse the cells three times with PBS.
-
121
Add 1 mL of fresh medium to the cells, and then observe and image the cells using an FV1000 laser scanning confocal microscope.
In vitro evaluation of gene silencing by siRNA NPs
Timing ~2 d
-
122
Seed 1 mL of HeLa-Luc cells in a 12-well plate at a density of 6 × 104 cells per well (complete RPMI 1640 medium). Incubate the cells at 37 °C in a humidified incubator containing 5% (vol/vol) CO2 for 24 h (or until 50–70% confluent).
-
123
Prepare 400 µL of siLuc NPs (S2P50 NPs; Dy647-siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2.)
-
124
Add 2.5, 5 and 10 µL of Dy647-siLuc NPs to three Eppendorf tubes (1.7 mL), respectively. Prepare all the samples in triplicates. Prepare a PBS control (or 0 nM siLuc) for the bioluminescence signal of cells treated with PBS.
-
125
Add 1 mL of RPMI 1640 medium to each of the tubes to dilute the NPs.
-
126
Mix the solution by pipetting gently.
-
127
Remove the cell medium, and add 1 mL of the medium containing Dy647-siLuc NPs (or the PBS control) to the cells (in triplicate). Final concentrations of Dy647-siLuc in the wells are 0, 12.5, 25 and 50 nM.
Critical step
For optimal gene silencing efficiency, make sure the cells are 50–70% confluent at the time of adding NPs.
-
128
Put the cells back in the incubator.
-
129
Remove the NPs after 24 h incubation. Add 1 mL of medium containing Steady-Glo Luciferase reagent (substrate) to the cells, and keep the cells in the incubator for 5 min.
Critical step
Steady-Glo Luciferase reagents can easily lose their activities through repeated freeze–thaw cycles; divide the reagent into aliquots, and always protect from light.
-
130
Determine the luciferase expression of the cells by measuring their bioluminescence signals using an In-Vivo Xtreme imaging system with a CCD camera.
-
131
Determine the gene silencing efficiency by quantifying the average radiance (photons per second per cm2 per steradian) within regions of interest using Bruker MI SE software.
In vitro viability assessment of siRNA NPs
Timing ~4 d
-
132
Seed 100 µL of HeLa-Luc (RPMI 1640 medium), RAW 264.7 (DMEM medium) and HEK-293 (EMEM medium) cells in three 96-well plates at a density of 5 × 103 cells per well, respectively. Incubate the cells at 37 °C in a humidified incubator containing 5% (vol/vol) CO2 for 24 h. Prepare a blank control (wells without cells) to measure the absorbance of medium without any cells. Prepare enough wells to run five replicates of each sample.
-
133
Prepare 400 µL of siLuc NPs (S2P50 NPs; siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2)
-
134
Add 2.5, 5, 10 and 20 µL of siLuc NPs to four Eppendorf tubes (2 mL), respectively. Add 2 mL of RPMI 1640 medium to each of the tubes to dilute the NPs. Prepare a PBS control to measure the absorbance of cells treated with PBS.
-
135
Mix the solution by pipetting gently.
-
136
Remove the cell medium, and add 100 µL of the medium containing siLuc NPs (or PBS control) to the wells of each plate. Prepare five replicate wells for each NP sample. The final concentrations of siLuc in the wells are 0, 6.25, 12.5, 25 and 50 nM, respectively.
-
137
Put the cells back in the incubator.
-
138
Remove the NPs after 24 h incubation. Add fresh medium to the cells, and incubate for another 48 h.
Critical step
The evaporation of cell medium in the outer wells of the 96 well plate is pronounced after 48 h incubation, which can influence the growth of cells, so we recommend using only the inner wells of the plate.
-
139
Remove the cell medium, and add 100 µL of medium containing alamarBlue cell viability and proliferation reagents (add 10 µL alamarBlue reagents to 90 µL of medium) to the cells.
-
140
Put the cells back in the incubator, and incubate for 1 h.
-
141
Determine the viability of the cells by measuring the absorbance at 570 nm using a plate reader.
-
142
Calculate the viability of NP-treated cells according to the following equation:
$$\% \,{{{\mathrm{Cell}}}}\,{{{\mathrm{viability}}}} = 100 \times \left( {{{{\mathrm{sample}}}} - {{{\mathrm{blank}}}}} \right)/\left( {{{{\mathrm{PBS}}}} - {{{\mathrm{blank}}}}} \right).$$
In vitro apoptosis assessment of siRNA NPs
Timing ~4 d
-
143
Seed 0.5 mL of RAW 264.7 (DMEM medium) cells in a 24-well plate at a density of 3 × 104 cells per well. Incubate the cells at 37 °C in a humidified incubator containing 5% (vol/vol) CO2 for 24 h.
-
144
Prepare 400 µL of siLuc NPs (S2P0 NPs, S2P50 NPs; siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2)
-
145
Add 20 µL of S2P0 NPs and S2P50 NPs to two Eppendorf tubes (2 mL), respectively. Add 2 mL of DMEM medium to each of the tubes to dilute the NPs. Prepare a PBS control to later measure the apoptosis of cells treated with only PBS.
-
146
Mix the solution by pipetting gently.
-
147
Remove the cell medium, and add 0.5 mL of medium containing siLuc NPs (or PBS control) to the cells. Prepare three replicates for each NP sample. The final concentration of siLuc in each well is 50 nM.
-
148
Put the cells back in the incubator.
-
149
Remove the NPs after 24 h incubation. Add fresh medium to the cells, and incubate for another 48 h.
-
150
Remove the cell medium, wash once with PBS, detach cells with trypsin, wash and collect the cells by centrifugation (1,000g, 4 °C, 5 min).
-
151
Double stain the cells with AV-FITC and PI (Annexin V-FITC Apoptosis Detection kit). Prepare a blank control: unstained cells. Prepare an AV-FITC control: cells stained with AV-FITC (no PI). Prepare a PI control: cells stained with PI (no AV-FITC).
Critical step
Keep the stained cells in dark or cover them with aluminum foil owing to the light sensitivity of AV-FITC and PI.
-
152
Determine the degree of apoptosis of the cells by flow cytometry. Analyze AV-FITC staining using the FITC signal detector and PI staining using the phycoerythrin emission signal detector. For the gating strategy for flow cytometry, first set a live gate (gate 1) in the forward scatter/side scatter plot to exclude cell debris. Then, use the cell populations from gate 1 for the downstream AV/PI display. Flow cytometry density plots show AV (X-axis) and PI (Y-axis) staining of cells. The right lower quadrant represents AV positive/PI negative staining indicating early apoptosis. The right upper quadrant represents both high AV and PI staining indicating late apoptosis. The left upper quadrant represents low AV and high PI staining indicating necrosis. The left lower quadrant indicates viable cells.
In vitro proliferation assessment of siRNA NPs
Timing ~8 d
-
153
Seed 2 mL of RAW 264.7 (DMEM medium) cells in a six-well plate at a density of 2 × 104 cells per well. Incubate the cells at 37 °C in a humidified incubator containing 5% (vol/vol) CO2 for 24 h. Prepare enough cell-seeded wells to run triplicate of each sample. Leave several wells empty as a blank control to later measure the absorbance of medium without cells.
-
154
Prepare 400 µL of siLuc NPs (S2P0 NPs, S2P50 NPs; siLuc 5 µM) according to the procedure for the preparation of NPs (Steps 31–52 using the formulations specified in Table 2.)
-
155
Add 80 µL of S2P0 NPs and S2P50 NPs to two Falcon tubes (15 mL), respectively. Add 8 mL of DMEM medium to each of the tubes to dilute the NPs.
-
156
Mix the solution by pipetting gently.
-
157
Remove the cell medium, and add 2 mL of medium containing siLuc NPs (or PBS control) to the cells. Prepare three replicates for each NP sample. The final concentration of siLuc in each well is 50 nM.
-
158
Put the cells back in the incubator.
-
159
Remove the NPs after 24 h incubation. Add fresh medium to the cells, and incubate for another 7 d.
-
160
At each timepoint (day 1, 2, 3, 4, 5, 6 and 7), remove the cell medium and add 2 mL of medium containing alamarBlue cell viability and proliferation reagents to the cells.
Critical step
Handle the alamarBlue reagents in dark owing to their light sensitivity.
-
161
Put the cells back to the incubator, and incubate for 1 h.
-
162
Determine the proliferation of the cells by measuring the absorbance at 570 nm using a plate reader.
-
163
Remove the medium containing alamarBlue reagents after each measurement, and add fresh medium to cells to allow the continued growth of the cells.
-
164
Calculate the proliferation of the cells according to the following equation:
$${}\% \,{{{\mathrm{Relative}}}}\,{{{\mathrm{cell}}}}\,{{{\mathrm{proliferation}}}} = 100 \times \left( {{{{\mathrm{sample}}}}_{{{{\mathrm{day}}}}\,{{{\mathrm{n}}}}} - {{{\mathrm{sample}}}}_{{{{\mathrm{day}}}}\,1}} \right)/ \left( {{{{\mathrm{sample}}}}_{{{{\mathrm{day}}}}\,1} - {{{\mathrm{blank}}}}} \right),{{{\mathrm{n}}}} = 1 - 7.$$
Treatment of WD-fed Ldlr −/− mice with S2P50 siCamk2g NPs
Timing ~4 weeks
-
165
Prepare 2 mL of S2P50 siCamk2g NPs and S2P50 siCtrl NPs (siRNA 5 µM) according to the procedure for the preparation of NPs (2 mL of NPs is required for single-dose injection for seven to nine mice). The siCtrl NPs is the control NPs prepared using nontherapeutic siLuc. Follow Steps 31–52 using the formulations specified in Table 2. This is fivefold-larger scale than reported in the above steps.
-
166
After establishing the mouse model of atherosclerosis (see ‘Reagent setup’), randomly divide the mice into three groups and inject intravenously two doses of PBS, S2P50 siCtrl NPs (1 nmol siRNA per injection, 200 µL) or S2P50 siCamk2g NPs (1 nmol siRNA per injection, 200 µL) per week to the mice, respectively. In each week, inject the second dose 3 or 4 d after the injection of the first dose. For statistical analysis, n = 7–9 mice per group are required per assay.
-
167
Maintain the mice on the WD during the treatment period.
-
168
After 4 weeks of treatment, euthanize the mice with CO2, collect the blood by left ventricular puncture and perfuse the vasculature with cold PBS.
-
169
Fix the heart with attached aortic roots in 4% (wt/vol) paraformaldehyde followed by paraffin embedding or treat it with optical cutting temperature compound and freeze it.
-
170
Isolate sections of the aortic root with sizes of 8 or 6 µm from the acetone-fixed/paraffin-embedded or frozen samples for the analysis of atherosclerotic lesions.
Silence of CaMKIIγ in lesional macrophages of WD-fed Ldlr −/− mice
Timing ~2 d
-
171
Block the obtained aortic root sections with 1% (wt/vol) BSA at room temperature for 1 h. For statistical analysis, n = 7–9 sections per group are required per assay.
-
172
Incubate the sections with primary anti-CaMKIIγ (1:400) and anti-Mac2 (1:10,000), or with anti-MerTK (1:500) at 4 °C overnight.
Critical step
To maintain the optimal activity of the primary antibody, handle the antibodies on ice.
-
173
Rinse the sections with PBS three times.
-
174
Incubate the sections with fluorescently labeled secondary antibodies for 2 h at room temperature.
-
175
After rinsing the sections with PBS three times, incubate the sections with DAPI for 20 min at room temperature.
Critical step
Keep the stained sections in the dark or cover them with aluminum foil owing to the light sensitivity of the dye-labeled antibodies and DAPI.
-
176
Rinse the sections (n = 7–9 sections per group) with PBS three times. Observe and image the sections using a Zeiss epifluorescence microscope, and analyze the images using FIJI software. The PBS-treated control is used to investigate gene silencing of sections from mice treated with PBS. The siCtrl NPs-treated control is used to investigate gene silencing of sections from mice treated with S2P50 siCtrl NPs.
Efferocytosis in lesional macrophages of WD-fed Ldlr −/− mice
Timing ~2 d
-
177
Incubate the acetone-fixed aortic root sections (from Step 170) with TUNEL staining reagents at 37 °C for 1 h. For statistical analysis, n = 7–9 sections per group are required per assay.
-
178
Wash the sections three times with PBS.
-
179
Block the sections with 1% (wt/vol) BSA at room temperature for 1 h.
-
180
Incubate the sections with primary anti-Mac2 (1:10, 000) at 4 °C overnight.
-
181
Rinse the sections with PBS three times.
-
182
Incubate the sections with fluorescently labeled secondary antibodies for 2 h at room temperature.
-
183
After rinsing the sections with PBS three times, incubate the sections with DAPI for 20 min at room temperature.
Critical step
Keep the stained sections in the dark or cover them with aluminum foil.
-
184
Rinse the sections with PBS three times. Observe and image the sections using a Zeiss epifluorescence microscope, and analyze the images using FIJI software.
-
185
Quantify the lesional efferocytosis as the ratio of TUNEL+ cells (red) in contact with Mac2+ macrophages (green) to isolated TUNEL+ cells. The PBS-treated control is used to investigate efferocytosis of sections from mice treated with PBS. The siCtrl NPs-treated control is used to investigate efferocytosis of sections from mice treated with S2P50 siCtrl NPs.
Plaque stability of WD-fed Ldlr −/− mice
Timing ~2 d
-
186
Stain the collagen of aortic root sections (from Step 170) with picrosirius red according to the manufacturer’s instructions. For statistical analysis, n = 7–9 sections per group are required per assay.
-
187
Observe and image the sections with light microscopy.
-
188
Obtain the necrotic core area by measuring the area from the images.
-
189
Quantify the fibrous (collagen) cap thickness from one midpoint and two shoulder regions of the plaque, and normalize it to the lesion size (Fig. 5g–j). The PBS-treated control is used to investigate plaque stability of sections from mice treated with PBS. The siCtrl NPs-treated control is used to investigate plaque stability of sections from mice treated with S2P50 siCtrl NPs.
In vivo evaluation of the toxicity of S2P50 siCamk2g NPs
Timing ~5 weeks
-
190
Purchase 6-week-old female BALB/c mice from Jackson Laboratory.
-
191
Conduct all the procedures under the protocols approved by the IACUC at your institution.
-
192
Prepare 800 µL of S2P50 siCamk2g NPs (siRNA 5 µM) according to the procedure for the preparation of NPs. Follow Steps 31–52 using the formulations specified in Table 2. This is twofold-larger scale than reported in the above steps.
-
193
Randomly divide the mice into two groups, and inject intravenously two doses of PBS or S2P50 siCamk2g NPs (1 nmol siRNA per injection, 200 µL) per week to the mice, respectively. For statistical analysis, n = 3 mice per group.
-
194
After 4 weeks of administration, euthanize the mice with CO2 3 d after the last administration; collect the blood by left ventricular puncture.
Critical step
After blood collection, immediately turn the blood collection tubes upside down a few times to mix blood with the anticoagulants.
-
195
Collect the major organs, and fix them in 4% (wt/vol) paraformaldehyde followed by paraffin embedding.
-
196
Isolate sections from the paraffin-embedded organs, statin them with H&E according to the manufacturer’s instructions.
-
197
Observe and image the sections with microscopy.
-
198
Evaluate various hematological parameters of the blood, including ALT, AST, BUN, HCT, Hb, LY, MCV, MCH, MCHC, NPC, RBC and WBC. PBS-treated control is used to investigate H&E staining of sections from mice treated with PBS, and hematological parameters of the blood from mice treated with PBS.
Troubleshooting
Troubleshooting advice can be found in Table 3.
Timing
-
Steps 1–14, synthesis of cationic lipid (G0-C14): 3 d
-
Steps 15–24, synthesis of ligand-modified lipid–PEG (DSPE–PEG–S2P): 3 d
-
Steps 25–30, preparation of siRNA stock solution: 1 h
-
Steps 31–52, preparation of siRNA-loaded NPs: 4 h
-
Steps 53–58, DLS characterization of siRNA NPs: 5 h
-
Steps 59–68, TEM characterization of siRNA NPs: 6 h
-
Steps 69–91, gel electrophoresis of siRNA NPs: 16 h
-
Steps 92–101, determination of the siRNA EE: 4h
-
Steps 102–108, evaluation of the stability of siRNA NPs: 3 d
-
Steps 109–121, characterization of the endosomal escape of siRNA NPs: 30 h
-
Steps 122–131, in vitro evaluation of gene silencing by siRNA NPs: 2 d
-
Steps 132–142, in vitro viability assessment of siRNA NPs: 4 d
-
Steps 143–152, in vitro apoptosis assessment of siRNA NPs: 4 d
-
Steps 153–164, in vitro proliferation assessment of siRNA NPs: 8 d
-
Steps 165–170, treatment of WD-fed Ldlr−/− mice with S2P50 siCamk2g NPs: 4 weeks
-
Steps 171–176, the silence of CaMKIIγ in lesional macrophages of WD-fed Ldlr−/− mice: 2 d
-
Steps 177–185, efferocytosis evaluation in lesional macrophages of WD-fed Ldlr−/− mice: 2 d
-
Steps 186–189, plaque stability evaluation of WD-fed Ldlr−/− mice: 2 d
-
Steps 190–198, in vivo evaluation of the toxicity of S2P50 siCamk2g NPs: 5 weeks
Anticipated results
Following this protocol, researchers will be able to synthesize the NP components, prepare the siRNA-loaded polymer–lipid hybrid NPs and characterize the NPs. We also describe the in vivo treatment of atherosclerosis in a mouse model as one of the applications of our polymer–lipid hybrid NPs. The results generated by this protocol can be anticipated based on our previous studies26,60. For the synthesis of G0-C14, a light-green solid with a yield of ~50% was achieved from 1.0 g of G0 starting material. The conjugation of DSPE–PEG–Mal (200 mg) with S2P (63.7 mg) produced a white powder (DSPE–PEG–S2P) with a yield of ~30%. The structures of G0-C14 and DSPE–PEG–S2P were confirmed by 1H NMR analysis, which can be found in Supplementary Fig. 1.
The physicochemical properties of the siRNA NPs were characterized by DLS and TEM. The nontargeted siRNA NPs (S2P0 NPs) had a diameter of 108 ± 2.8 nm (intensity size distribution) and a zeta potential of −7.43 ± 0.94 mV (Fig. 4a,b). As expected, the conjugation of S2P ligand to the NPs resulted in targeted S2P50 NPs with a slightly increased diameter of 116.2 ± 2.5 nm (intensity size distribution) and zeta potential of −2.66 ± 1.01 mV. TEM images revealed that both the nontargeted (S2P0 NPs) and targeted (S2P50 NPs) NPs had spherical morphologies (Fig. 4d). It is worth noting that both S2P0 and S2P50 NPs had similar siRNA EE of ~50% (Fig. 4c). The stability of the siRNA NPs in biological medium is critical to their long blood circulation, which allows the accumulation of siRNA NPs at lesional sites. We demonstrated that the S2P0 and S2P50 NPs were stable in PBS solution containing 10% (vol/vol) serum at 37 °C for at least 3 d as evidenced by the negligible changes in their particle size (Supplementary Fig. 2a, b). We further confirmed that the S2P50 NPs could efficiently protect the siRNA from enzymatic degradation when incubated in serum solution (Supplementary Fig. 2c). Importantly, our siRNA NPs were able to escape from the endosomes after 4 h incubation with HeLa-Luc cells as demonstrated by the confocal microscopy images (Supplementary Fig. 3).

a,b, Size (a) and zeta potential (b) of siRNA NPs (S2P0 and S2P50) determined by DLS; n = 3. c, siRNA EE of S2P0 and S2P50 NPs measured by plate reader. Dy647-siRNA was loaded into the NPs (n.s., not significant); n = 6. d, TEM images of S2P0 and S2P50 NPs. e, In vitro gene silencing of siRNA NPs. HeLa (HeLa-Luc) cells were incubated with S2P50 NPs containing varying concentrations of siLuc for 24 h, and their luciferase-expressing was characterized by bioluminescence imaging. f, The average radiance of the cells was quantified; n = 3. Data are presented as mean ± SEM. ****P < 0.0001 on one-way ANOVA. a–f adapted with permission from ref. 26, American Association for the Advancement of Science.
We then evaluated the gene silencing effect of siRNA NPs in HeLa-Luc cells. Our siLuc-loaded S2P50 NPs were able to effectively silence the Luc expression of HeLa-Luc cells at a siLuc concentration as low as 12.5 nM as demonstrated by the bioluminescence imaging (Fig. 4e,f). In addition, the in vitro biocompatibility of the siRNA NPs was assessed by different assays. The HeLa-Luc, RAW 264.7 and HEK-293 cells treated with S2P50 NPs containing varying concentrations of siLuc (6.25-50 nM) displayed high cell viabilities of >80% (Supplementary Fig. 4a). The treatment of RAW 264.7 cells with S2P0 and S2P50 NPs (siLuc 50 nM) induced minimal cell apoptosis comparable to that induced by PBS (Supplementary Fig. 4b). Both of the S2P0 and S2P50 NPs (siLuc 50 nM) did not show any significant inhibitions to the growth of RAW 264.7 cells after monitoring their proliferation as long as 7 d (Supplementary Fig. 4c).
Next, we assessed the in vivo applications of our polymer–lipid hybrid NPs using the treatment of atherosclerosis in a mouse model as an example. To this end, we first established a mouse model of atherosclerosis by feeding the Ldlr gene-deficient (Ldlr-/-) mice with a high-fat WD for 8 weeks. The resulting WD-fed Ldlr−/− mice were intravenously injected with either PBS, S2P50 siCtrl NPs or S2P50 siCamk2g NPs. After the treatment, the sections of the aortic roots of the mice were isolated for analysis of the therapeutic effect of NPs. The immunofluorescence imaging revealed that the S2P50 siCamk2g NPs could efficiently silence the expression of the CaMKIIγ in the lesional macrophages, resulting in the activation of the MerTK pathway (Fig. 5a–d). TUNEL assay showed that the S2P50 siCamk2g NPs-induced activation of the MerTK pathway could substantially promote the lesional efferocytosis (Fig. 5e,f). Consequently, plaque stability was improved in the S2P50 siCamk2g NP-treated groups as evidenced by the smaller necrotic area and thicker fibrous cap compared with that of the PBS or S2P50 siCtrl NP-treated groups (Fig. 5g–j).

Eight-week WD-fed Ldlr−/− mice were injected intravenously with two doses of PBS, S2P50 siCtrl NPs (1 nmol siRNA per injection) or S2P50 siCamk2g NPs (1 nmol siRNA per injection) per week for 4 weeks; n = 7–9 mice per group. a, Sections of the aortic root were immunostained for CaMKIIγ and Mac2. Nuclei were stained with DAPI. Scale bars, 200 µm. b, The mean fluorescence intensity (MFI) for CaMKIIγ of each group was normalized to the PBS group. c, The MFI for MerTK of each group was normalized to the PBS group. d, Sections of the aortic root were immunostained for MerTK. Nuclei were stained with DAPI. Scale bars, 200 µm. e, TUNEL assay was used to characterize the apoptotic cells (ACs) of sections of the aortic root. Arrows indicate isolated or free TUNEL+, cells, and arrowheads indicate TUNEL+ cells in contact with Mac2+ macrophages. Scale bars, 50 µm. f, The lesional efferocytosis was quantified as the ratio of TUNEL+ cells (red) in contact with Mac2+ macrophages (green) to isolated TUNEL+ cells. g, Sections of the aortic root were stained with picrosirius red to determine the lesion size and necrotic core area. The necrotic core area is quantified. h, Microscopy images of the sections. Scale bars, 200 µm. i, Percent necrotic core area of lesion area is quantified. j, The fibrous (collagen) cap thickness is quantified from one midpoint and two shoulder regions of the plaque and normalized to the lesion size. *P < 0.05 on one-way ANOVA. Data are presented as mean ± SEM. a–j adapted with permission from ref. 26, American Association for the Advancement of Science. All the animal procedures were conducted in accordance with the protocols reviewed and approved by IACUC at Harvard Medical School and Columbia University Irving Medical Center.
We also investigated the in vivo toxicity of our polymer–lipid hybrid NPs under the same conditions (e.g., dose, frequency) as the assay for therapeutic effect evaluation. To this end, BALB/c mice were injected intravenously with PBS or S2P50 siCamk2g NPs for 4 weeks, two doses per week. After the treatment, sections of the major organs and blood of the mice were isolated and collected for analysis of the toxicity of NPs. No obvious damage to the major organs of NP-treated mice was observed as indicated by the H&E staining images (Fig. 6a). In addition, blood from both PBS- and NP-treated mice showed no significant differences in a wide range of hematological parameters, including ALT, AST, BUN, HCT, Hb, LY, MCV, MCH, MCHC, NPC, RBC and WBC (Fig. 6b).

Six-week-old female BALB/c mice were injected intravenously with two doses of PBS or S2P50 siCamk2g NPs (1 nmol siRNA per injection) per week for 4 weeks; n = 3 mice per group. The mice were euthanized by CO2 inhalation 3 d after the last administration. a, The major organs were sectioned, followed by H&E staining. Scale bars, 100 µm. b, Blood was collected, and various hematological parameters were evaluated, including ALT, AST, BUN, HCT, Hb, LY, MCV, MCH, MCHC, NPC, RBC and WBC. Data are presented as mean ± SEM. The differences between PBS and S2P50 siCamk2g NPs groups are not statistically significant for all parameters (Student’s t-test). a and b adapted with permission from ref. 26, American Association for the Advancement of Science. All the animal procedures were conducted in accordance with the protocols reviewed and approved by IACUC at Harvard Medical School and Columbia University Irving Medical Center.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data discussed in this protocol are available in the supporting primary research papers (https://doi.org/10.1126/scitranslmed.aay1063) and https://doi.org/10.1073/pnas.1303958110). The raw datasets are available for research purposes from the corresponding authors upon reasonable request.
References
Schultze, J. L., Schmieder, A. & Goerdt, S. Macrophage activation in human diseases. Semin. Immunol. 27, 249–256 (2015).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Ross, R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340, 115–126 (1999).
Kuznetsova, T., Prange, K. H. M., Glass, C. K. & de Winther, M. P. J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17, 216–228 (2020).
Huang, X. et al. Intercalation-driven formation of siRNA nanogels for cancer therapy. Nano Lett. 21, 9706–9714 (2021).
Zhu, X. et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc. Natl Acad. Sci. USA 112, 7779–7784 (2015).
Davidson, B. L. & McCray, P. B. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 12, 329–340 (2011).
Whitehead, K. A., Langer, R. & Anderson, D. G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 8, 129–138 (2009).
Castanotto, D. & Rossi, J. J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 457, 426–433 (2009).
Chen, J. et al. Synthesis and use of an amphiphilic dendrimer for siRNA delivery into primary immune cells. Nat. Protoc. 16, 327–351 (2021).
Loynachan, C. N. et al. Renal clearable catalytic gold nanoclusters for in vivo disease monitoring. Nat. Nanotechnol. 14, 883–890 (2019).
Terrazas, M. & Kool, E. T. RNA major groove modifications improve siRNA stability and biological activity. Nucleic Acids Res. 37, 346–353 (2008).
Sioud, M., Furset, G. & Cekaite, L. Suppression of immunostimulatory siRNA-driven innate immune activation by 2′-modified RNAs. Biochem. Biophys. Res. Commun. 361, 122–126 (2007).
Choung, S., Kim, Y. J., Kim, S., Park, H.-O. & Choi, Y.-C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 342, 919–927 (2006).
Kanasty, R., Dorkin, J. R., Vegas, A. & Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 12, 967–977 (2013).
Yin, H. et al. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541–555 (2014).
Mullard, A. FDA approves landmark RNAi drug. Nat. Rev. Drug Discov. 17, 613–614 (2018).
Frank-Kamenetsky, M. et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl Acad. Sci. USA 105, 11915–11920 (2008).
Zimmermann, T. S. et al. RNAi-mediated gene silencing in non-human primates. Nature 441, 111–114 (2006).
Tadin-Strapps, M. et al. siRNA-induced liver ApoB knockdown lowers serum LDL-cholesterol in a mouse model with human-like serum lipids. J. Lipid Res. 52, 1084–1097 (2011).
Pan, H. et al. Anti-JNK2 peptide–siRNA nanostructures improve plaque endothelium and reduce thrombotic risk in atherosclerotic mice. Int. J. Nanomed. 13, 5187 (2018).
Flores, A. M. et al. Nanoparticle therapy for vascular diseases. Arterioscler. Thromb. Vasc. Biol. 39, 635–646 (2019).
Sager, H. B. et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci. Transl. Med. 8, 342ra380–342ra380 (2016).
Dahlman, J. E. et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol. 9, 648 (2014).
Leuschner, F. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 29, 1005–1010 (2011).
Tao, W. et al. siRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci. Transl. Med. 12, eaay1063 (2020).
Doran, A. C. et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Investig. 127, 4075–4089 (2017).
Doran, A. C., Yurdagul, A. & Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 20, 254–267 (2020).
Finn, A. V., Nakano, M., Narula, J., Kolodgie, F. D. & Virmani, R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 30, 1282–1292 (2010).
Islam, M. A. et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2, 850–864 (2018).
Kong, N. et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 11, eaaw1565 (2019).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Tang, Z. et al. A materials-science perspective on tackling COVID-19. Nat. Rev. Mater. 5, 847–860 (2020).
Wang, Z. et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature 592, 616–622 (2021).
Bradley, T. et al. Antibody responses after a single dose of SARS-CoV-2 mRNA vaccine. N. Engl. J. Med. 384, 1959–1961 (2021).
Tang, Z. et al. Insights from nanotechnology in COVID-19 treatment. Nano Today 36, 101019 (2021).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol. Ther. 27, 710–728 (2019).
Ahmadi, S. et al. Stimulus-responsive sequential release systems for drug and gene delivery. Nano Today 34, 100914 (2020).
Liu, Y. & Chen, X. Macrophage-targeted gene therapy to improve the atherosclerotic plaque stability. Matter 3, 621–622 (2020).
Zhao, W., Hou, X., Vick, O. G. & Dong, Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials 217, 119291 (2019).
Wang, Y. & Yu, C. Emerging concepts of nanobiotechnology in mRNA delivery. Angew. Chem. Int. Ed. 59, 23374–23385 (2020).
Vaughan, H. J., Green, J. J. & Tzeng, S. Y. Cancer‐targeting nanoparticles for combinatorial nucleic acid delivery. Adv. Mater. 32, 1901081 (2020).
Zhou, Y. et al. Blood–brain barrier-penetrating siRNA nanomedicine for Alzheimer’s disease therapy. Sci. Adv. 6, eabc7031 (2020).
Truong, N. P. et al. An influenza virus-inspired polymer system for the timed release of siRNA. Nat. Commun. 4, 1902 (2013).
Hu, Q., Li, H., Wang, L., Gu, H. & Fan, C. DNA nanotechnology-enabled drug delivery systems. Chem. Rev. 119, 6459–6506 (2019).
Hu, Q., Wang, S., Wang, L., Gu, H. & Fan, C. DNA nanostructure-based systems for intelligent delivery of therapeutic oligonucleotides. Adv. Healthc. Mater. 7, 1701153–1701172 (2018).
Lee, H. et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 7, 389–393 (2012).
Thai, H. B. D. et al. Kidney-targeted cytosolic delivery of siRNA using a small-sized mirror DNA tetrahedron for enhanced potency. ACS Cent. Sci. 6, 2250–2258 (2020).
Bujold, K. E., Hsu, J. C. C. & Sleiman, H. F. Optimized DNA “nanosuitcases” for encapsulation and conditional release of siRNA. J. Am. Chem. Soc. 138, 14030–14038 (2016).
Rahman, M. A. et al. Systemic delivery of bc12‐targeting siRNA by DNA nanoparticles suppresses cancer cell growth. Angew. Chem. Int. Ed. 56, 16023–16027 (2017).
Semple, S. C. et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 28, 172–176 (2010).
Kulkarni, J. A., Witzigmann, D., Chen, S., Cullis, P. R. & van der Meel, R. Lipid nanoparticle technology for clinical translation of siRNA therapeutics. Acc. Chem. Res. 52, 2435–2444 (2019).
Whitehead, K. A. et al. Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity. Nat. Commun. 5, 4277 (2014).
Wittrup, A. et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 33, 870–876 (2015).
Sahay, G. et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 31, 653–658 (2013).
Liu, S. et al. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR–Cas gene editing. Nat. Mater. 20, 701–710 (2021).
Cheng, Q. et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 15, 313–320 (2020).
Xu, X. et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc. Natl Acad. Sci. USA 110, 18638–18643 (2013).
Wilson, D. S. et al. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 9, 923–928 (2010).
Díez, S., Miguéliz, I. & De Ilarduya, C. T. Targeted cationic poly (d, l-lactic-co-glycolic acid) nanoparticles for gene delivery to cultured cells. Cell. Mol. Biol. Lett. 14, 347–362 (2009).
Shi, J., Xiao, Z., Votruba, A. R., Vilos, C. & Farokhzad, O. C. Differentially charged hollow core/shell lipid–polymer–lipid hybrid nanoparticles for small interfering RNA delivery. Angew. Chem. Int. Ed. 50, 7027–7031 (2011).
Mui, B. L. et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. Nucleic Acids 2, e139 (2013).
Love, K. T. et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl Acad. Sci. USA 107, 1864–1869 (2010).
Schöttler, S. et al. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 11, 372–377 (2016).
Petros, R. A. & DeSimone, J. M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 9, 615–627 (2010).
Moghimi, S. M., Hunter, A. C. & Murray, J. C. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol. Rev. 53, 283–318 (2001).
Gilleron, J. et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 31, 638 (2013).
Ahmed, S. A., Gogal, R. M. Jr & Walsh, J. E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H] thymidine incorporation assay. J. Immunol. Methods 170, 211–224 (1994).
Miller, L. R. et al. Considering sex as a biological variable in preclinical research. FASEB J. 31, 29–34 (2017).
Fredman, G. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 7, 275ra220–275ra220 (2015).
Zhu, X. et al. Surface de-PEGylation controls nanoparticle-mediated sirna delivery in vitro and in vivo. Theranostics 7, 1990–2002 (2017).
Liu, J. et al. Fast and efficient CRISPR/Cas9 genome editing in vivo enabled by bioreducible lipid and messenger RNA nanoparticles. Adv. Mater. 31, 1902575 (2019).
Ensuring accurate resource identification. Nat. Protoc. 15, 1879–1880 (2020).
Acknowledgements
This study is supported by the American Heart Association (AHA) Collaborative Science Award (No. 2018A004190, to W.T.), Harvard Medical School/Brigham and Women’s Hospital Khoury Innovation Award (No. 2020A003219, to W.T.), Anesthesiology Department Basic Scientist Grant (No. 2420 BPA075, to W.T.), Center for Nanomedicine Research Fund No.2019A014810, to W.T.) and National Institutes of Health (HL127464, HL145228 and HL087123, to I.T.; R00 HL145131; to A.Y.). W.T. also received a start-up package (for 3 years) from the Department of Anesthesiology, Perioperative and Pain Medicine to establish his independent research laboratory at Harvard Medical School and Brigham and Women’s Hospital.
Author information
Authors and Affiliations
Contributions
Conceptualization, W.T., I.T., A.Y.J; writing and editing, W.T., I.T., A.Y.J, X.H., C.L., N.K., Y.X.; all the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Quanyin Hu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Tao, W. et al. Sci. Transl. Med. 12, eaay1063 (2020): https://doi.org/10.1126/scitranslmed.aay1063
Zhu, X. et al. Proc. Natl. Acad. Sci. 112, 7779-7784 (2015): https://doi.org/10.1073/pnas.1505629112
Islam, M. A. et al. Nat. Biomed. Eng. 2, 850–864 (2018): https://doi.org/10.1038/s41551-018-0284-0
Kong, N. et al. Sci. Transl. Med. 11, eaaw1565 (2019): https://doi.org/10.1126/scitranslmed.aaw1565
Supplementary information
Supplementary Information
Supplementary Figs. 1-4.
Rights and permissions
About this article

Cite this article
Huang, X., Liu, C., Kong, N. et al. Synthesis of siRNA nanoparticles to silence plaque-destabilizing gene in atherosclerotic lesional macrophages. Nat Protoc 17, 748–780 (2022). https://doi.org/10.1038/s41596-021-00665-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00665-4
This article is cited by
-
Nobiletin alleviates atherosclerosis by inhibiting lipid uptake via the PPARG/CD36 pathway
Lipids in Health and Disease (2024)
-
Thylakoid engineered M2 macrophage for sonodynamic effect promoted cell therapy of early atherosclerosis
Nano Research (2024)
-
Engineering siRNA therapeutics: challenges and strategies
Journal of Nanobiotechnology (2023)
-
Inflammation and atherosclerosis: signaling pathways and therapeutic intervention
Signal Transduction and Targeted Therapy (2022)
-
The landscape of mRNA nanomedicine
Nature Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
