
Abstract
Mass-spectrometry-based proteomic analysis is a powerful approach for discovering new disease biomarkers. However, certain critical steps of study design such as cohort selection, evaluation of statistical power, sample blinding and randomization, and sample/data quality control are often neglected or underappreciated during experimental design and execution. This tutorial discusses important steps for designing and implementing a liquid-chromatography–mass-spectrometry-based biomarker discovery study. We describe the rationale, considerations and possible failures in each step of such studies, including experimental design, sample collection and processing, and data collection. We also provide guidance for major steps of data processing and final statistical analysis for meaningful biological interpretations along with highlights of several successful biomarker studies. The provided guidelines from study design to implementation to data interpretation serve as a reference for improving rigor and reproducibility of biomarker development studies.
Similar content being viewed by others
Main
More than 20,000 diseases have been reported to affect humans1, of which only a small portion are supported by accurate, sensitive and specific diagnostic tests. Even for diseases with well-established diagnostic assays, such as diabetes, the discovery of new prognostic biomarkers can enable further studies on disease development and progression. For example, type 1 diabetes mellitus can be diagnosed by measuring blood glucose concentration, but the disease is known to be preceded by immunological changes sometimes years before clinical manifestation. Biomarkers for detecting and discriminating early stages of the disease could contribute to an improved understanding of the associated etiology and pathogenicity, while informing new therapies and prevention targets2,3. Additionally, biomarkers are urgently needed to improve many current diagnostic assays, particularly in the context of personalized medicine, such as for inflammatory bowel disease4. There is also a demand for biomarkers that can predict the outcome of the patient or that can be used in clinical trials to follow the progression of patients to treatments5. In this context, proteomic analysis of biological samples, including tissues, blood plasma, exhaled breath condensate, saliva and urine, are promising approaches for discovering new biomarkers and advancing knowledge of disease pathology, prevention, diagnostics and therapeutics across a wide range of diseases.
Proteomic analysis of human biofluids and tissues can detect and quantify thousands of proteins, leading to the discovery of many potential biomarkers. However, improper experimental design, lack of standardized procedures and quality controls (QCs) (see Box 1 for key terminology) for sample collection and analyses, and failure to validate identified biomarkers have led to reproducibility challenges and identification of biomarkers that are not clinically relevant6,7,8,9,10,11,12. There are some excellent reviews highlighting the main issues faced during biomarker development8,9,10,12,13,14. Indeed, experimental rigor and reproducibility have been the theme of ample discussion in the scientific community. Funding and regulatory agencies and scientific journals have implemented guidelines to these aspects of research15,16,17,18,19. A systematic review of 7,631 tuberculosis biomarker citations revealed some common challenges that cause misinterpretation: (1) small number of samples (underpowered studies), (2) inappropriate control groups, and (3) overemphasizing P-values for candidate discovery without further validation efforts20. The authors also found that most of these studies failed to specify whether the study was performed in a blinded fashion20.
In this tutorial, we describe key points that should be considered for performing biomarker discovery experiments based on liquid-chromatography–mass-spectrometry analysis of human clinical samples. Experimental rationale, possible failing points and QC considerations are provided for sample selection criteria, sample preparation, data collection and data analysis. These recommendations are based on protocols developed by our group and by colleagues from NIH-funded consortia that we participate in, such as Clinical Proteomic Tumor Analysis Consortium (CPTAC), The Environmental Determinants of Diabetes in the Young (TEDDY), Molecular Transducers of Physical Activity Consortium (MoTrPAC), Early Detection Research Network (EDRN), Cancer Moonshot and Undiagnosed Diseases Network (UDN). Overall, careful implementation of each of these steps should enhance the rigor and reproducibility of biomarker studies and the overall likelihood of discovering relevant, actionable biomarkers.
Phases of biomarker development
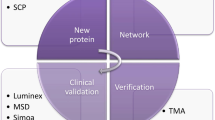
Biomarker development is typically described in the literature as being divided into three phases: discovery, verification and validation (Fig. 1)21,22. The validation phase is itself often divided into two stages: analytical validation and clinical validation, with the latter often described as ‘qualification’. Here we will focus only on the analytical aspects of biomarker validation. Fewer peptides and proteins are measured and more samples and subjects are studied as the study moves from discovery to verification to validation phases22,23]. This transition requires a different set of quality assessments to ensure the analytical validity of an assay. In general, analytical validity includes several standard parameters including precision, specificity, sensitivity, recovery and stability. Precision includes a measure of repeatability, which refers to within-day variability, and reproducibility, which refers to day-to-day variability24. Repeated measurements can be used to define an assay’s coefficient of variation under different conditions and at different concentrations. The robustness of a coefficient of variation must be interpreted within the context of what is a clinically significant change in the analyte. As part of the validation of reproducibility, it is also important to test whether an assay produces similar results when performed by different individuals and in different laboratories.

Biomarker discovery is usually divided into three different phases: discovery, verification and validation. In the discovery phase, a small number of samples is submitted for in-depth proteomics analysis where thousands of proteins are measured to identify biomarker candidates. Often, larger cohorts of samples are analyzed in the subsequent phases, increasing the statistical power. Biomarker candidates are also downselected each developmental phase based on their performance to accurate predict the disease or condition. In some cases, a combination rather than individual protein is tested as a biomarker. In the verification phase, biomarker candidates undergo additional proteomics analysis to verify both their identities and expression in the same or similar samples as in the discovery phase. A few of the most promising candidates are tested in the validation phase to determine its performance for clinical use.
The discovery phase is focused on the identification of a large number of candidate biomarkers. This phase is primarily based on in-depth, untargeted proteomic analysis to identify and quantify as many proteins as possible21,25, leading to the identification of tens to hundreds of biomarker candidates that will then be assessed further in the verification and validation phases. However, due to the cost, logistics and relatively low throughput of discovery proteomics, this phase is often carried out using a limited number of samples. Because the discovery phase involves the putative (yet still highly confident) identification of peptide (and therefore protein) markers based on matching experimental tandem mass (MS/MS) spectra to computationally predicted MS/MS spectra, the initial identifications must be verified in the same or similar samples as used for the discovery phase.
The verification phase is focused on confirming that the abundances of target peptides are significantly different between disease and control groups compared through quantitative measurements. Stable-isotope-labeled, synthetic peptides are often spiked into the samples of interest to facilitate confident detection and quantification of targeted peptides using targeted mass spectrometry (MS)-based assays. The confident detection of the putative markers is determined by coelution and similarity of MS/MS fragment pattern compared with the synthetic peptide standards26. Subsequent steps of the fold change verification are usually carried out across clinical samples. Targeted MS provides much more accurate quantitative measurement of biomarker candidates with relatively high analytical throughput19,23,27. The number of samples analyzed in this phase depends on the complexity of the disease condition, prior research and the analytical assay platform. It should be determined by power analysis, but often dozens to hundreds of samples are analyzed to confirm the differential abundances of the biomarker candidates.
The goal of the analytical validation phase is to confirm the utility of the biomarker assays by analyzing samples from an expanded or independent cohort of individuals that have the same disease as was investigated in the discovery and verification phases. This provides a measure of robustness of the biomarkers and of the assays used to measure them. Usually, only a few (three to ten) of the best biomarker candidates are tested in the analytical validation phase. There are, however, many conditions where panels containing multiple biomarkers have better diagnostic performance28,29. Therefore, it is important to consider how many candidates need to be evaluated. Similar to the verification phase, the number of samples should be determined by power analysis and depends on multiple factors, including the number of candidate biomarkers used. It can vary from tens to thousands of samples from patients in an appropriate clinical patient cohort. This phase is often performed by either immunological assays, such as ELISA, if available, or targeted MS assays in cases where specific antibodies are not available. If both the verification and analytical validation phases are done using targeted MS, these phases will have the same design and experimental considerations, so for the purposes of this tutorial we have combined the considerations of both of these phases below.
Subject selection
Critical to making appropriate inference in disease biomarker prediction is selection of samples representative of both disease cases as well as the population from which the cases are drawn30. The limited number of samples that can be analyzed in the different phases reinforces the importance of properly selecting the study cohort. Sample matching improves the comparative analysis and reduces the number of samples required to obtain proper statistical power. However, this needs to be done carefully as it limits inference to a generalizable population, and the process of matching itself may preclude the ability to evaluate the direct effect of any of the matched characteristics because the sampling scheme is inherently biased31,32,33. Samples from subjects with disease should be appropriately paired with those from nondiseased individuals with similar characteristics for comparison to reduce confounding factors. Many diseases are differentially affected by sex, age, body mass index, race/ethnicity, comorbidities and preexisting conditions. Therefore, such factors should be considered during experimental design, and testing and control groups should be matched as closely as possible during cohort recruitment. Additional samples or comparison groups might be needed to account for multiple factors or outcomes of the disease due to these covariates. Conventional observational studies may use a number of different approaches for study design, such as secondary assay or analysis of clinical trials, cohort, nested case–cohort, case–control, or others (see Box 2 for details on different types of study design), with different degrees of bias34,35,36 in case and control sample selection inherent to each design. Modern statistical methods, such as inverse probability weighting37,38 or Bayesian methods39, should be used to adjust estimates of effect or estimate the degree to which selection bias may influence the findings. Further consideration for making appropriate inference is the problem of confounding factors40, which should be typically addressed either by randomization in experimental studies or adjustment in observational ones, although the problem of residual confounding41 can persist in both circumstances.
Once the cohort is selected, the study should be approved by an institutional review board or equivalent before the project starts. An institutional review board reviews protocols, consent forms and captured information to assure that the rights and welfare of the human subjects (sample donors) are protected.
Power analysis
The number of study subjects and associated samples is dependent on the selected study design, which is itself dependent on the scientific question and intended inference42. In this context, a power analysis provides an estimate of the number of study subjects and associated samples required to obtain statistical significance for a certain effect size. For binary outcomes, the effect size is typically a fold change, but for more complicated designs with multiple treatment groups or longitudinal samples, the effect size is set by the goals of the experiment to be low or high, dependent on the level of effect that needs to be detected. This is akin to a larger sample size being required to detect a twofold change versus a threefold change.
For biomarker studies, one must consider both the epidemiological and analytical factors that influence the required number of study subjects. The incidence of disease in the general population, likely attrition rate and biological variability in protein expression levels will impact the number of individuals needing to be recruited. The inherent analytical variability in the proteomics platform to be used for biomarker discovery will also contribute to the final cohort size.
Case–control or nested case–cohort studies are approaches that can be taken to reduce the population size required for analysis; this is especially useful in situations where you would want to collect a large amount of data for each individual—something that would be very difficult to achieve in a classical cohort study. These designs trade cost for improvements in statistical power43,44, with a design focused on the outcome of interest.
Cohort studies track the incidence of diseases or conditions across a temporal sequence, which can take longer but provide better capacity for strong causal inference. This type of study often requires larger sample sizes for the same statistical power45, and focuses on the exposures of interest.
It is sometimes convenient to perform secondary analysis of trials (i.e., querying for different disease outcomes or factors that were not the main question of the study) or intervention studies, but some caution should be exercised. Often studies are sufficiently large and well powered for the primary analysis46, but the secondary analyses may require statistical adjustment to correct for confounding factors, making the study underpowered. It is therefore important to have a statistical analysis plan for both the primary and the secondary analysis in place before performing the power analysis.
Power analysis is more complicated in studies where the analysis involves simultaneously measuring multiple analytes, because standard approaches to compute power are based on a single metric of estimated variance, irrespective of the study design. Even in the same set of MS runs, different peptides have different variability and require different numbers of samples for proper statistical power. To manage this issue, the standard approach is to estimate the variances of all proteins from a proteomics study where data were collected within a similar population and sample matrix47,48,49, then select a threshold based on the minimum percentage of proteins to be quantified. In this context, the threshold is the statistical power expected for the majority of the proteins. This threshold is rarely 100% because variances tend to be highly skewed across an omics-based dataset, especially for low-intensity peptides/proteins. A few proteins with extreme variability in either expression or measurability can drive up the sample size dramatically. For example, Levin et al. showed that for a study to be properly powered at a minimum of 80% (or 0.8), with a detectable fold change of 1.5 comparing two groups for all proteins, the minimum sample size is 60 per group47. Reducing the power expectation to 75% of the proteins results in a minimum sample size of 35, and reducing the power requirement even further to 50% decreases the minimum number of samples per group to 16. This will come with the tradeoff that fewer proteins will be adequately powered for the comparison of interest. Therefore, it is important to evaluate during the experimental design the tradeoff of the number of proteins that will be properly powered for a given sample size and detectable fold change based on the needs of the study.
As an example of power calculation for a large-scale MS analysis, the Metabolomics Core for the NIH Common Fund Undiagnosed Diseases Network (UDN) Phase I evaluated the number of samples from healthy individuals required for building a baseline of metabolite and lipid reference values to be compared against similar profiles from individuals with disease. In the UDN, each patient had a unique and undiagnosed illness; therefore, it was important to have a well-defined baseline of normal metabolite and lipid profiles to compare against an N of 1. Using data from previous analyses of similar samples, the minimum numbers of reference samples were selected on the basis of power calculations considering a Student’s t-test with a type I error of 0.05 and a twofold detectable change for 80% of the tested molecules. It was found that 102 samples would be necessary for urinary metabolomics, and 136 samples for plasma lipidomics50. In another example, a proteomics study on the mechanism of pancreatic β-cell killing by proinflammatory cytokines found that only four samples would be necessary for a twofold detectable change using Student’s t-test with a type I error of 0.05 for 80% of the proteins51. These examples show that the number of required samples can be drastically different. This difference depends on the biological and technical variability and the study design.
Sample handling, collection, storage and tracking
Both discovery and validation efforts can be impacted by a number of preanalytic variables that should be carefully considered when designing sample collection protocols and when deciding the characteristics of clinical cohorts for sample collections. Analysis may be influenced by physiologic factors, including age, sex, body mass index, fasting status, timing of collection (i.e., circadian or diurnal influences), phase of menstrual cycle, exercise status, season of collection, medical comorbidities and interfering medications52,53,54,55,56,57. Due to this biological variability, it is important to keep the experimental/analytical variance to a minimum to obtain meaningful data. The impact of these variables can be minimized by strict matching criteria for prospective collections and through development and implementation of standard operating procedures (SOPs) by those responsible for sample collection. SOPs should include detailed criteria for sample collection and processing, and whenever possible, manufacturers and lots of reagents should remain consistent for the duration of a study58. Results may be influenced by the type of anticoagulant used in blood collection tubes or by the type of collection tube used for other biofluids59. Certain labile analytes may require specific additives such as protease inhibitors or antioxidants for stabilization60. To avoid sample degradation, the time between sample collection, sample processing and number of freeze–thaw cycles should be minimized and also kept consistent among all samples to avoid introduction of artifacts in the data. Of note regarding sample preservation, extensive efforts have been dedicated to evaluating the suitability of formalin-fixed paraffin-embedded (FFPE) samples for proteomics analysis61,62. These studies have demonstrated that, when combined with specialized sample preparation protocols discussed further below, FFPE specimens are well suited to biomarker discovery studies63,64.
When preparing the sample collection, questionnaires should be formulated to capture all the relevant metadata, including sex, age, height, weight, race/ethnicity, comorbidities and preexisting conditions. Depending on the disease or condition under study, it is also important to capture information about any prescribed medicines or diets, as they can impact the composition of the collected sample. For instance, even a meal has a strong effect on the composition of the plasma proteome65. Once the protocol is approved and the SOP is established, the samples should be collected in a standardized way, taking care to prevent degradation (low temperature or addition of proper preservatives). Sample accessioning (i.e., assigning accession numbers) should be performed with care to avoid mislabeling, and the use of barcoding and printing labels rather than hand-writing can be employed to minimize the chances of sample mix-up66.
Once the samples are collected, storing them in a single batch provides an opportunity to control for variability in how the researcher handles the samples. Different peptides/proteins might have different stability based on their physical/chemical properties67. Therefore, freeze–thaw cycles should be minimized, and long-term storage should be done at −80 °C. Stability of the samples can be tested by spiking internal standards and monitoring their abundances across different freeze–thaw cycles and storage time. Such experiments can also provide information on analyte recovery and assay specificity and sensitivity68. Caution should be used when analyzing previously collected samples, especially where details of collection and storage are not available and when combining samples from multiple sources58. These factors can introduce variability in the data.
The importance of sample blinding
Technical bias in assay-based studies can present an additional source of error69. Small differences in sample handling and preparation throughout the experiment can cause major differences in the results and compromise the integrity of the study. Therefore, when it is possible, samples should be randomized and deidentified by the statistician, with no subject information given to researchers who will process and analyze the samples, to avoid inadvertent differences in sample handling based on some subject feature, such as case status. Additionally, attention should be paid to assessing and minimizing, if possible, batch effects when the number of samples exceeds the assay batch size. One approach is to randomize cases and controls across chip or plate locations, to avoid batch clustering based on assay chip or plate, date, or reagent. There are some situations where blinding is not feasible, e.g., when samples have identifiable characteristics (different color, sizes, texture, etc.). Other cases where it is difficult to perform completely blind studies are studies that involve either food or surgery, where both the subjects and researchers know the control and treatment groups70. When blinding is impractical, analyzing samples from additional independent cohorts helps to confirm that biomarker candidate identification was not due to human bias71,72.
Considerations for discovery-phase experiments
The main goal of the discovery phase is to analyze as many biomarker candidates as possible. To achieve this goal, an in-depth proteomics analysis is carried out by liquid chromatography (LC)-MS/MS with a limited number of samples, with a focus on the depth of proteome coverage. Depending on the sample complexity, abundant protein depletion and peptide prefractionation is performed to increase the chances of detecting proteins present in low abundance. In addition, peptide labeling with isobaric tags can be used for multiplexing several samples in a single experiment, which decreases variability between measurements. Checkpoints along with QCs and statistical analysis improve the chance of identifying meaningful biomarker candidates. The overall workflow is shown in Fig. 2, while checkpoints, expected results, potential pitfalls and troubleshooting are listed in Table 1.

The main consideration points for each step of the workflow are shown. Note that an example for blood plasma analysis is shown, but other sample types may have some additional or fewer steps in the workflow. For tissue analysis, the immunodepletion step should be replaced by a tissue lysis step, the details of which are documented in the text.
Abundant protein depletion
Blood plasma and serum are challenging specimens because of their complex composition and the presence of highly abundant proteins. The most abundant plasma protein, serum albumin, is present at 35–50 mg/mL in normal conditions, whereas cytokines are only present in low pg/mL range, differing by a factor of 1010. In addition, the 20 most abundant proteins account for 97% of the total plasma protein mass73. These highly abundant proteins represent a major challenge for proteomic analysis since the MS data collection is biased towards high-abundance peptides74. Two main approaches have been taken: immunodepletion and fractionation by chromatography.
The removal of highly abundant proteins through immunodepletion allows for better detection of moderate- and low-abundance proteins75,76. Unfortunately, immunodepletion can also codeplete other associated proteins77. Other methods to simplify sample complexity, such as denaturing size exclusion chromatography or extensive high-pH reversed-phase fractionation, have been successfully applied78, with the trade-off of an increased number of LC-MS/MS runs. Therefore, the method of decreasing sample complexity needs to be considered carefully.
Immunodepletion has to be performed before protein digestion. If this approach is chosen, we recommend that you run a QC sample before each batch of samples to be depleted. Consistently running QCs of well-characterized samples, such as NIST 1950 plasma, allows the development of baselines for determining fluctuations in instrument and depletion column performance. This can be monitored with UV detection and overlaying the elution profiles. For instance, an increase in the unbound protein peak might represent degradation of the column or improper buffer pH. Samples should be kept at low temperatures (i.e., on ice or at 4 °C) to avoid proteolytic degradation.
Removal of abundant proteins or peptides by chromatographic fractionation is discussed further below as part of the information relating to the chromatographic separations.
Protein digestion
Sample preparation for proteomic analysis typically includes the initial homogenization of solid samples, protein solubilization, and lysis, followed by enzymatic digestion and solid phase extraction to remove contaminants (Table 2). We have previously found that protein extraction is a major source of experimental variability79. Therefore, it needs to be performed in the most consistent way possible. Lysis buffers usually consist of a buffering agent (e.g., ammonium bicarbonate, Tris-HCl or triethylammonium bicarbonate) and denaturing agents (e.g., urea, guanidine hydrochloride, thiourea). They are formulated and optimized to release and improve solubility of proteins by disrupting hydrogen bonds and hydrophobic interactions between and within proteins. When working with FFPE specimens, harsher extraction conditions are required to undo the extensive protein crosslinking that occurs during fixation80,81,82. It may also be necessary to start with larger specimens when working with FFPE tissue, to ensure sufficient protein amounts for downstream processing. Reduction of protein disulfide bonds (with dithiothreitol, tris(2-carboxyethyl)phosphine) and alkylation of the free SH-groups (with iodoacetamide, iodoacetic acid, acrylamide or chloroacetamide) improves sample digestion and MS detection of cysteine-containing peptides83. Lysis buffer may contain protease and other inhibitors (e.g., phosphatase inhibitors for phosphopeptide analysis) to minimize the biodegradation of extracted proteins. Protease inhibitors should be carefully chosen to not interfere with the protein digestion step.
Performing protein quantification on the cell lysate is an important step to ensure the extraction efficiency, calculation of enzyme needed for sample digestion and allowing control checks of the following steps. This procedure also allows normalization of the digest parameters through the study, and it is essential for the final quality of the digest and the protocol reproducibility. For protein digestion, trypsin has been considered as the gold standard in proteomics sample preparation, but other enzymes such as endoproteinases Glu-C and Lys-C can also provide additional information. Walmsley et al. have shown that trypsin from different sources can add substantial variability to the samples84. Therefore, it is important to use enzyme from the same lot throughout the experiment. The experimental conditions for trypsin digestion can be adjusted for a specific application. Typically, trypsin digestion is performed at neutral pH at 37 °C, and it may take up to 18 h. The digestion is stopped by reducing the pH of the sample with trifluoroacetic or formic acid. The acidification of the samples also allows for better performance on the sample desalting step and better recovery of the peptides85. Sample desalting using solid-phase extraction is vital since it removes salts and buffers that are not compatible with the following steps. At this point, quantification of the peptides should be performed to assess the recovery of the samples and ensure that variability between samples are in a reasonable range. As an additional QC step, a small aliquot of digested peptides can be taken at this point and analyzed by 1D LC-MS/MS analysis to interrogate digestion quality and identify problematic samples prior to subsequent steps.
Peptide labeling with isobaric tags and sample multiplexing
There are multiple approaches for quantitative global proteomics analysis, all with advantages and disadvantages14. Peptide labeling with isobaric tags (e.g., tandem mass tag (TMT) reagents) has become a popular method in large-scale discovery studies because it allows in-depth proteome coverage with sample multiplexing to achieve relatively good throughput and reduced technical variability86,87, enabling the discovery of low-abundance biomarker candidates. The disadvantage of isobaric labeling is that these approaches often lead to underestimation of fold changes between samples due to interfering signals coming from reagent impurities, background noise and cofragmented peptides87. On the other hand, label-free analysis by data-dependent acquisition or data-independent acquisition provide more accurate fold changes. One disadvantage of the label-free approach is that only one sample can be analyzed at a time, compared with up to 16 in the TMT experiments. Compared with TMT-labeled experiments, data-dependent acquisition and data-independent acquisition analyses often lead to low coverage of the proteome in challenging samples, such as plasma and serum88,89, since TMT-labeled samples are more amenable to fractionation prior to LC-MS/MS. Prefractionation of data-dependent acquisition and data-independent acquisition samples adds the challenge of increasing the analysis time and may introduce more variability to the samples. Despite all these approaches being powerful and successfully used for global proteomics analysis90,91,92,93,94, in this section, we will mainly cover isobaric tag labeling because of its popularity and the complexity of overall workflow.
To facilitate the comparison between multiple sets of TMT experiments, a ‘universal’ reference sample can be included in one of the multiplexing channels for each TMT set. This reference sample can be just an aliquot mixture of all the samples. It can be used to normalize signal intensities across different TMT sets and also serves as a standard for QC analysis. There are two important steps in peptide labeling and multiplexing: (1) ensure the right pH of the samples since it affects the efficiency of peptide derivatization, and (2) quantify peptides before labeling and multiplexing. We have found that remaining acids from solid phase extractions can lower the pH of the samples, drastically reducing the efficiency of TMT labeling. We have also observed that post hoc data normalization is effective for only small variations of sample loading. A postlabeling QC is also recommended. To achieve this, a small aliquot is taken from each sample prior to quenching the labeling reaction, mixed, and analyzed by LC-MS/MS to determine the efficiency of labeling for each channel. Because the labeling reaction is left unquenched, samples with low labeling efficiency can often be effectively rescued by adding additional label.
Peptide-level fractionation
Digestion of tissue lysates, whole cells or body fluids can generate >500,000 peptides per sample95. In shotgun proteomics, the depth of the analysis is partially limited by the tandem mass spectra scan rates. Therefore, reducing the complexity of the sample by prefractionating the peptides improves the proteomic coverage95. Peptide fractionation prior to the LC-MS/MS analysis also helps with the problem of ratio compression. Ratio compression refers to a phenomenon where the measured fold changes are smaller than the real abundance differences present in the samples, and is a known issue in experiments where peptides are labeled with isobaric tags. This problem is caused by cofragmentation of multiple coeluting peptides (and anything else that would create a high chemical background) such that the peak contains reporter ion fragments from both the selected peptide and these interfering factors87. Prefractionation of peptides results in a lower chemical background and better separation of peptides from each other, reducing the ratio compression issue96.
There are several types of chromatography that can be used for peptide prefractionation, including strong-cation exchange, hydrophilic interaction and reverse phase (reviewed in reference97). High-pH reverse-phase separation has become increasingly popular as the first dimension for tryptic peptide fractionation in a biomarker discovery workflow. For large projects, assay variables should be as consistent as possible, i.e., buffers, columns, gradients and temperatures of separation, to have the most reproducible measurements. Indeed, even small fluctuations in pH can lead to major shifts in retention times98. Monitoring elution profiles with UV detection also helps to ensure that the separation is reproducible. For preservation of sample quality, peptides are stored dry in vials to be rehydrated prior to LC-MS/MS analysis.
Data collection
Many parameters must be monitored for the LC-MS/MS data collection to be effective. Calibrations should also be performed following mass spectrometer manufacturer recommendations to ensure the accuracy of the measurements. The performance of the instrument should be assessed by regularly running well-characterized standard samples. For a robust assessment of the instrument performance, the standard samples should have similar complexity and properties to the samples to be analyzed. The mass spectrometers should be serviced when the analysis of standard samples indicates suboptimal performance, which is determined by comparing with the historical performance of the instrument (e.g., a QQ or Bland–Altman plot). For instance, in our laboratory, we use the tryptic digest of the bacterium Shewanella oneidensis as the standard sample. However, each laboratory can develop their own QC sample based on material availability. There are several QC standards from bacterial and mammalian cells, as well as human biofluids, commercially available. The analysis of this standard sample on a high-resolution mass spectrometer such as Q-Exactive (Thermo Fisher Scientific) with a 100 min chromatography gradient usually leads to the identification of ~12,000 peptides. We clean the instrument once these numbers drop below 11,000 identified peptides, which restores the number of identifications (Fig. 3). Peak width and other metrics can also give indication of specific problems with the LC or the mass spectrometer99. Therefore, it is important to set baselines for multiple parameters to assess the overall performance of the instrument. Samples should be blocked and randomized when analyzed to avoid bias due to instrument performance decay100,101. Our data and those from other groups have shown that even normal decay in instrument performance can introduce confounding factors to the data101,102. Standards should run before and after a block of samples. The block size is determined considering mass spectrometer performance drift over time and separation length. This allows breaks between blocks to clean, calibrate and perform preventative maintenance. Randomization should be done within blocks. Complete randomization can lead to imbalances (i.e., more control samples run first and more of the test samples run after, or vice versa), which can reintroduce some confounding factors101. Without blocking, data collection would need to be restarted from the beginning to avoid bias due to the instrument performance differences before and after servicing.

In our laboratory, we use a tryptic digest of the bacterium Shewanella oneidensis as a standard sample to check the LC-MS/MS performance. This standard is run before and after each batch of samples. a, Number of identified peptides in S. oneidensis runs. Note a slow decay in the number of identified peptides, which is almost unnoticeable in consecutive runs but has a major effect across time. The number of peptide identifications was reestablished after cleaning the instrument. b,c, Chromatograms from analysis of S. oneidensis before and after instrument cleaning, respectively. This shows the cumulative reduction in instrument performance across time.
Data QC
The quality of the sample and data is crucial for obtaining meaningful results. Therefore, in our protocol, we implement QC measurements for each major procedure step. Quantification of proteins and peptides is a good way to assess whether a sample is being lost during depletion, digestion and labeling steps. During the crucial period of data collection, it is desirable to assess the quality of data acquired in real time. Relatively few tools have been developed for real-time monitoring of LC-MS data quality. We recently introduced the Quality Control Analysis in Real Time (QC-ART) software, a tool for evaluating data as they are acquired to dynamically flag potential issues with instrument performance or sample quality102. QC-ART identifies local (run-to-run variations) and global (across large sets of data) deviations in data quality due to either biological or technical sources of variability. For instance, QC-ART can detect trends in signal intensity decline or reduction in the number of identified peptides, which can result from instrument performance decay102. Chromatographic shifts, especially in the first and last quartile of the elution time, may represent problems in column integrity, solvent composition or tubing dead volumes. The QC-ART procedure is similar to that of Matzke et al.103 in the context of the statistical outlier algorithm employed but adds a dynamic modeling component to analyze the data in a streaming LC-MS environment.
In addition to real-time monitoring tools, several QC methods exist for checking data postcollection to remove low-quality data that would degrade downstream statistics (reviewed in reference104). Data QC allows the detection of important differences in the samples that might not result from drifts in instrument performance or problem in sample preparation. For instance, QC-ART was able to detect minor differences in chromatography profiles between samples, with reduction of some peak intensities but appearance or increase of others (see highlighted region of Fig. 4a). A deeper investigation led to the identification of oxidation in amino acid residues (Fig. 4b), such as cysteine, tryptophan and tyrosine (Fig. 4c,d), which, despite being previously described, were underappreciated during analysis of plasma samples. By recognizing and specifically searching for these oxidations, the proteome coverage was significantly improved (P < 0.05) (Fig. 4e,f)102. Therefore, QC not only identifies technical issues, but can also lead to the identification of characteristics of the samples that are different across the cohort, such as posttranslational modifications.

a, Total-ion chromatogram from analysis of three LC-MS/MS runs from corresponding high-pH reversed-phase chromatography fractions of different multiplexed sets of isobaric-tagged samples. The runs were analyzed by QC-ART, and the flagged run is highlighted. The highlighted region has a different peak profile compared with the unflagged runs. b, A selected m/z range of the region highlighted in a. The analysis reviewed a shift of 15.99 Da, corresponding to the mass of an oxidation, on the peptide GQYCYELDEK, which does not contain the methionine residues, which are commonly searched during peptide identification. c, Workflow of the MSGF+ database searches to identify new oxidized residues. The searches considered oxidation in any residue and used Ascore163 to ensure the site of modification. d, Normalized counts of oxidized amino acid residues. e,f, Average number of peptide (e) and protein (f) identifications per fraction of reanalyzed data. The blue bars represent the database search performed considering methionine oxidation as the only possible modification, whereas the red bars also considered methionine, cysteine, tryptophan and tyrosine oxidations. This shows that not only can QC analysis find runs with drift in in sample preparation and instrument performance, but it can also find runs that have distinct profiles due to unexpected posttranslational modifications. The asterisks represent P ≤ 0.05 by t-test. Reproduced from ref. 102 with permission from the American Society for Biochemistry and Molecular Biology.
Data analysis
Currently, there are excellent tools for peptide identification, such as MS-GF+, MSFragger, Andromeda and TagGraph105,106,107,108. Although most of these tools work in an almost completely automated fashion, an important aspect of the peptide identification is to control the number of false-positive identifications. The most common approach is to use a target-decoy database for sequence searching, which allows calculation of the false-discovery rate (FDR)109. Most commonly, FDRs are kept at 1% at the protein and peptide levels to maximize the balance between rigor in peptide identification and yield of biological information. Less-stringent FDRs can introduce a substantial number of false-positive identifications, while more stringent FDR criteria may exclude biologically relevant peptides. The balance of these choices will depend on the scientific question, and whether it is preferable in the study context to identify more false positives or more false negatives. Manual inspection of the spectra can also be performed, but it is only practical for small numbers of peptides since it is labor intensive and requires well-trained personnel. For instance, in our laboratory, we only manually inspect spectra from posttranslationally modified peptides that we use to study signaling mechanisms. True-positive peptides usually have sequentially matching tandem mass fragments110. In addition, the tandem mass analysis of some posttranslational modifications generates diagnostic fragments that can be used to further confirm their presence. For subsequent targeted proteomics experiments, peptides will also be validated in the verification/validation phases using their heavy labeled internal standard versions.
Once a set of peptides is identified, their intensity information is extracted for the quantitative analysis. In the first quantification step, normalization is focused on accounting for the bias introduced due to technical and biological variation. Common normalization strategies include total abundance normalization to the average or median, linear-regression-based approaches, quantile normalization and variance stabilization normalization (Vsn)111,112,113,114 (Table 3).
Despite these considerations, there is no consensus in the community on a single best strategy to normalization, and the optimal approach can vary based on sample type, study scale and the complexity of the sample matrix (e.g., cell lines, tissue, plasma). For example, global-based normalization makes two assumptions that might not hold115: (i) that the amount of peptide detected is proportional to the amount of protein present and (ii) that the total concentration of protein within all samples in an experiment is constant.
If the biological effect of a condition is to increase (or decrease) the total amount of protein produced in the sample, or generate different types of proteins resulting in a change in the relationship between total proteins and peptides quantified, then global normalization strategies would introduce bias. Examples of this are conditions where the abundance of inflammatory proteins is at a level where lower-abundance proteins are no longer detectable in the analysis.
Webb-Robertson et al.113, proposed a strategy called Statistical Procedure for the Analyses of peptide abundance Normalization Strategies (SPANS), which performs multiple normalizations and uses metrics of variability and bias to make recommendations. More recently, Valikangas et al.114 noted that the number of methods available in SPANS is limited and performed a comprehensive review of multiple normalization approaches. They found that Vsn was the most effective for reducing variation between technical replicates and performed well for evaluation metrics associated on differential expression statistics. The goal of Vsn normalization is to bring the samples to the same scale by first performing a transformation to remove variance caused by systematic experimental factors and then, second, apply a generalized log2 transformation. Since Vsn is focused on addressing the relationship between the variance and mean intensity for the example data used by Valikangas et al., it also underestimates the log2 fold changes of spiked in proteins. Supervised approaches to incorporate more accurate estimates of variance also show great promise in managing the differences in measured protein across samples116,117. These approaches use machine learning algorithms, mostly random forest and support vector machines, to identify and quantify batch effects or other systematic experimental factors, from which they adjust for these effects. The primary issue with this approach currently is that the accuracy of these approaches for smaller datasets has not been well quantified. In general, most guidance regarding normalization of proteomics data suggests careful consideration of both data and scientific goals of the analysis in order to select the most appropriate method.
Statistical analysis is generally performed in a univariate manner, evaluating each protein independently using an appropriate test based on the experimental design. For discrete outcomes, standard approaches such as a standard t-test, ANOVA or the generalized linear mixed-effects model (GLMM) are the usual approaches in order of experimental complexity. For example, in a simple bench biology experiment of a cell line, a simple t-test may be adequate, but in a complex analysis with multiple levels of a factor or multiple experimental parameters, an ANOVA would be well suited. Further, in complex cohort studies where repeated measures of subjects may be taken or other covariates, such as age, need to be adjusted for, a GLMM is a flexible strategy to perform statistics. However, in some cases, nonparametric equivalents of these tests should be utilized if the underlying assumptions of the model are not met (e.g., a standard t-test yields meaningful information only if the distribution of the data is normal; if the distribution is not normal, then one could use a Wilcoxon rank sum test). Quantitative outcomes are most commonly evaluated using linear- and nonlinear-regression-based approaches.
Proteomic experiments generate a large number of peptides/proteins, and each are evaluated independently using one of the tests previously described (e.g., ANOVA, Wilcoxon rank sum test). This yields a large number of test statistics (P-values), for which the standard type 1 error used to draw a significance threshold is no longer accurate and an approach must be taken to obtain a more accurate measure of the uncertainty or error level. This is commonly referred to as an FDR calculation. There are many approaches to perform this task, such as a Bonferroni correction, which simply defines a protein as significant if the P-value is less than 0.05/P, where P is the total number of proteins statistically analyzed118. This is one of the most conservative approaches to adjusting for this error. Alternatively, there have been multiple methods developed to control the FDR, such as Benjamini and Hochberg, Strimmer, and q-values, the latter of which is probably the most widely used119,120. In general, these approaches perform a correction based on an estimate of the ratio of false positives to true positives at a defined test statistic (P-value), which is estimated from the data.
It should be noted that the utilization of FDR calculations is extremely challenging for specific experimental designs, such as ANOVA and GLMM when testing multiple factors or time-based factors. Thus, it is not unusual to evaluate the data generated in the discovery phase using multiple type 1 error thresholds, sorting, machine learning121,122 or network-based123,124 inference to identify the best candidates for targeted analyses.
Considerations for experiments of the verification and validation phases
Verification and validation phases for selected biomarker candidates from discovery phase are mostly performed with targeted MS-based assays or targeted proteomics analysis26,125,126. Targeted proteomics is a complementary technique, where candidate biomarker peptides are measured alongside heavy-isotope-labeled synthetic counterparts. This not only improves the quantification process but also ensures that the correct peptide is being measured with high level of specificity. Selected-reaction monitoring (SRM, also known as multiple reaction monitoring) on a triple quadrupole mass spectrometer and parallel reaction monitoring on a high-resolution mass spectrometer (e.g., Q-Exactive) are commonly applied targeted MS techniques. In general, targeted MS assays provide high accuracy, selectivity and sensitivity, because they use two-stage mass filtering of both precursor and fragment ions with high resolution. Recent advances in MS have made it possible to perform large-scale candidate biomarker validation involving hundreds of peptides127,128,129.
Similar to the discovery phase, the validation phase has an extensive workflow from sample selection to assay development and data collection, to final data analysis (Fig. 5). Checkpoints, expected results, potential pitfalls and troubleshooting are listed in Table 1.

The main consideration points for each step of the workflow are shown.
Biomarker candidate prioritization
Biomarker discovery studies can lead to the identification of hundreds to thousands of candidates. Unfortunately, logistics and cost often limit the number of biomarker candidates that can be studied in the following verification and validation experiments. There is no community consensus on how candidates should be prioritized, and several strategies have been described, including prioritization based on statistical significance, machine learning analysis, functional-enrichment analysis, correlation with published literature, and integration of multi-omics datasets. Frequently, the main criteria for prioritizing biomarker candidates are their statistical significance and fold change when comparing cases versus controls130.
Machine learning approaches are powerful methods to prioritize biomarker candidates based on their performance in predicting the disease outcome131. A suite of machine learning techniques, such as logistic regression, random forests and support vector machines have been used to build predictive models of disease; however, the true power of this approach is in the identification of a multivariate biomarker panel. Various approaches, such as random forest feature importance metrics132 are common, as well as Bayesian integration and statistical sampling strategies that can be used to extract feature sets from disparate datasets121. While machine learning has been shown to be effective for selecting candidates, other more basic analyses, such as linear regression, can be as effective in many cases. For instance, Carnielli et al. have successfully verified biomarker candidates selected based on their association with the clinical characteristics of the patient, using linear regression94. Functional-enrichment analysis can also provide insights about the disease or condition and is applicable to lists of biomarkers identified either by univariate statistics or machine-learning-based biomarker discovery. This type of analysis allows the user to determine pathways that are likely to be altered in disease. Often, proteins from the same pathway have similar regulation; depending on the purpose of the study, you could purposefully choose protein candidates that represent different pathways (diversity of effect) or study those that are involved in the same pathway (mechanistic insight). Information from the literature can be very helpful, since a better understanding of the disease process can allow for the selection of more meaningful biomarker candidates, such as key regions of pathways (e.g., regulatory members and bottlenecks). Finally, a powerful approach is the integration of data from multi-omics measurements, which can select biomarkers that have positive correlations between their levels of transcript and proteins, for example, or enzymes and metabolites133.
Targeted peptide selection
After candidate prioritization, multiple peptides per protein are selected based on their detectability and SRM suitability. Suitable peptides for SRM assays typically need to be 6–25 amino acids in length, fully tryptic and without any missed cleavage sites (lysine and arginine before proline, KP/RP, are not considered missed cleavage)134. Peptides with different chemical properties (molecular weight, amino acid composition, length and hydrophobicity) should be included because peptides with similar characteristics will coelute. The duty cycle of the instrument limits the number of peptides that can be monitored simultaneously. Therefore, selecting targets across the length of the chromatographic separation, for example, with a retention time prediction tool135, allows maximization of the number of targeted peptides. Coelution can also cause signal interference between multiple peptides. Rost et al. developed a tool named SRMCollider that predicts interference between peptides and can be used to exclude problematic transitions136. Some amino acids have properties that are not ideal for developing assays. Methionine, asparagine and glutamine residues are prone to spontaneous modification into oxidized methionine, aspartate and glutamate, respectively134. Sequences containing these amino acids should be avoided. In addition, some sequences are hard to chemically synthesize137; analysis requires that you have a corresponding heavy-isotope-labeled standard, so one should choose a sequence that is easy to synthesize.
In deciding which standards to make, we recommend analysis of the alkylated version of cysteine-containing peptides (e.g., carbamidomethylation), because free cysteine residues can oxidize or dimerize into disulfide bonds. For the standard peptides, carbamidomethylated cysteine can be directly incorporated during synthesis.
All the candidate peptides need to be searched against the human proteome to ensure their uniqueness. In general, at least three unique peptides per protein should be selected at this stage as some peptides are excluded during assay development because of interfering signals or poor detectability.
LC-SRM assay development
Once the biomarker peptides have been chosen, LC-SRM assays are developed in three main steps: transition selection, gradient optimization and best peptide selection.
Transition selection
The importance of the first step is to choose transitions that are both specific and sensitive. Initially, five or six transitions per precursor ions are selected for developing the targeted proteomics assays based on their intensity in the tandem-mass spectra138. Some peptides may have more than one precursor ion, depending on the distribution of charge states. Next, stable-isotope-labeled peptide standards are spiked into a nonhuman peptide matrix (e.g., bacterial lysate, bovine serum albumin or chicken plasma digests) in multiple concentrations and analyzed by LC-SRM. The different concentrations of spiked standard peptides help to differentiate the actual signal versus the background. The best precursors and transitions are determined based on the highest signal intensity and least interference. A final number of two to four transitions per peptide are usually included in the assay. In addition, the collision energy can be optimized for individual transitions to further improve the sensitivity. This feature is available in Skyline, a popular software used for LC-SRM analysis139.
Optimize the LC gradient
In experiments measuring hundreds of peptides, it is crucial to have a well-balanced gradient. Peptides should not be aggregating in a narrow window of retention time. Instead, they should be well distributed across the entire gradient length. This will make it possible to schedule more transitions without a decrease in dwell time and sensitivity. Selection of peptides with distinct characteristics, as mentioned above, helps to distribute the peptides across the length of the gradient. Once the gradient is optimized, the last assay development step is to select peptides with the best performance.
Choose the best peptides
The best performing peptides are the ones that have good endogenous detectability, little matrix interference, and good correlation between peptides representing the same protein. This can be accessed by spiking the stable-isotope-labeled peptide standards in a set of test samples and monitoring the performance of all the peptides in an LC-SRM study. In general, at least one to two peptides per protein are included in the final targeted proteomics assay.
Assay evaluation
The sensitivity of the assay can be accessed by the limit of quantification (LOQ) and limit of detection (LOD) for peptides. There are three approaches to obtain the LODs and LOQs: (1) reverse response curve of increasing concentrations of stable-isotope-labeled internal standard peptides with endogenous peptides as reference, (2) forward calibration curve of increasing concentrations of unlabeled peptides in a matrix without the targeted proteins, and (3) a matrix-matched calibration curve approach by diluting sample matrix and a pooled reference matrix of diverged species at various ratios140. Additional characterization experiments can also be conducted, including the evaluation of repeatability, selectivity, stability and reproducible detection of endogenous analytes141.
Sample preparation
Biomarker validation studies have many similarities, with important considerations discussed above for discovery studies and some additional considerations to accommodate the increased throughput required to sufficiently expand the patient cohort. Our approach to increasing sample processing throughput has been to carry out the procedure in multiwell plates79. Targeted proteomics measurements require less sample input and fewer preparation steps, making it feasible to carry out preparation in commercially available 96-well plates.
Working in plate format requires some modifications to standard laboratory practices to maintain uniform application of SOPs across larger sample batches. First, when making reagent additions, the use of liquid handling robots is highly recommended, to increase both the speed and accuracy. Adding reagent to 96 or 192 wells using a single-channel pipette will introduce substantial differences in treatment conditions between sample 1 and sample 192. Furthermore, having a large number of repetitive tasks in a workflow makes it more prone to intermittent errors, such as missed samples, which will result in outliers and lost patient measurements from the study. Secondly, we have found that the largest contributor to sample variance in our plate-based sample preparation is nonuniform temperature during sample incubations79. Due to the geometry of the 96-well plate, samples in inner wells can experience a different temperature than those in outer wells. For this reason, it is critical to evaluate temperature distribution, for your incubator and chosen deep well plate. Lastly, QC for large processing batches is required to gain an accurate estimation of the variance across the entire study, which may take place over the course of years. To do this, we recommend the creation of a pooled sample containing aliquots from existing patients in the study, whenever possible. This sample is then included in multiple randomized positions on each well plate and carried through the entire analysis process142. In addition to determining variance, these samples serve as instrument QCs for maintaining optimal assay performance.
Stable-isotope-labeled standard peptide spiking and storage
In LC-SRM analysis, samples are spiked with heavy-isotope-labeled versions of each targeted peptide. To create consistent samples for SRM analysis, it is important to normalize the protein concentration using a suitable assay such as the bicinchoninic acid (BCA) assay. Adjusting all samples to the same concentration serves the dual purpose of creating more-stable light-to-heavy ratios for data analysis, and ensures the consistent sample loading necessary for reproducible chromatography. For projects with large cohort of samples, it is important to plan for enough stable-isotope-labeled standard peptide mixtures to use during the study of the entire cohort. Standard peptide mixture is often prepared in acidified solution, such as 0.1% formic acid in water with 15–30% acetonitrile. The mixture is prepared into aliquots in multiple vials, and each vial is enough for all the samples in a 96-well plate. The mixture aliquots are stored in a −80 °C freezer until their further usage67.
Immunoaffinity enrichment
Peptide immunoaffinity enrichment is a technique often coupled with targeted MS for improving the detection and quantification of low-abundance peptides. In this approach, heavy-isotope-labeled peptides are spiked into samples prior to enrichment, and they are captured along with their endogenous counterparts by specific antibodies143,144,145,146,147,148. This procedure decreases the overall sample complexity, boosting the signal of the targeted peptides. A few checkpoints in this approach are to ensure equal spiking of peptides and antibodies to the samples, and to ensure the correct pH for optimal capture143. Crosslinking antibodies to the beads can reduce the amount of these molecules in the samples and reduce the chemical background noise of the analysis143.
Data QC
The day-to-day QC and quality assurance (QA) in data acquisition can be quite overwhelming for a targeted proteomics study of thousands of samples. A graphical-user-interface-based software tool, Q4SRM149, can be used to rapidly access the signal from all stable-isotope-labeled standard peptides once the data acquisition is done and flags those that fail QC/QA metrics.
Data analysis
For LC-SRM data analysis, we usually use Skyline software139. Raw files were imported into Skyline along with peptide transitions. Normally, it is done in batch mode; for example, data files processed in the same 96-well plate can be imported and processed in one single Skyline file. Manual inspection of the data is often required to ensure the correct peak assignment and peak boundaries. While going through the manual inspection in Skyline, it is a good idea to inspect both graphs of retention time and peak area of individual peptides over all the samples to check any unusual behaviors. The total peak area ratio of endogenous peptides over stable-isotope-labeled internal standard peptides can be exported directly from Skyline for downstream analysis.
Establishing the robustness of the targeted MS assays
For large-scale validation phase using targeted MS assays, it is critical to fully characterize assays for each surrogate peptide for its performance to ensure the robustness of these assays in such applications. Recently, the Clinical Proteomic Tumor Analysis Consortium (CPTAC) and other groups have published assay characterization guidelines for ensuring robustness of the assays67,150,151,152. These guidelines recommend the following items:
-
(1)
Response curve: assays should be checked against a sample with similar complexity. For example, assays for human plasma analysis can be checked in chicken plasma, which has similar complexity but different peptides. This allows determination of the LOD and LOQ, and if the assay has a linear dose–response curve.
-
(2)
Selectivity: assays should be analyzed without internal standards and with low and medium concentrations (based on the linear curve) with multiple biological replicates to determine their selectivity.
-
(3)
Stability: the stability of peptides can be tested by spiking samples with internal standards and assessing the peak area variability after storage in different storage conditions (4, −20 and −80 °C), over time (weeks to months), and through free–thaw cycles.
-
(4)
Repeatability and reproducibility: assays can be tested by preparing and analyzing representative samples multiple times independently in different days.
These recommendations should be taken into close consideration before implementing assays for large-scale validation efforts. Once the assays are fully characterized, SOPs should be established for implementation.
Examples of successful biomarker studies
All successful biomarker studies involve multidisciplinary teams of clinicians, analytical chemists and statisticians. They require rigorous experimental design, considering potential technical issues and adequate numbers of samples.
To highlight the technical aspects described in this tutorial, we discuss a few examples of successful MS-based biomarker studies using different analytical pipelines (Table 4).
Type 1 diabetes
Zhang et al.153 performed a biomarker study comparing serum from individuals with type 1 diabetes to controls. The discovery experiment consisted of ten pooled sera from individuals with type 1 diabetes compared with controls of healthy individuals; each pool consisted of five individuals. Samples were depleted of 12 abundant proteins, digested with trypsin and analyzed by LC-MS. The analysis resulted in the identification of 24 differentially abundant proteins, which were verified by LC-SRM analysis of sera from 50 individuals with type 1 diabetes versus 100 healthy controls. The peptides were further examined in a third blind cohort of 10 individuals with type 1 diabetes versus 10 healthy controls, and against a cohort of 50 individuals with type 1 diabetes paired against 50 individuals with type 2 diabetes to test the biomarker performance to distinguish between the two diabetes forms. The study identified platelet basic protein and C1 inhibitor, both achieving 100% sensitivity and 100% specificity. Of these proteins, C1 inhibitor was particularly good in discriminating between the two types of diabetes153.
Oral squamous cell carcinoma
In a study of oral squamous cell carcinoma, Carnielli et al. explored the histopathological features to identify biomarkers94. In this type of cancer, morphological features, such as the invasive tumor front and the inner tumor region, are good indicators of the disease prognosis154. Therefore, they performed proteomics of laser capture microdissected tissue from 20 samples taken from each of six regions: small neoplastic island (abnormal tissue growth), large neoplastic island, and stroma from both invasive tumor front and inner tumor. Biomarker candidates were verified by immunohistochemistry (IHC) and were prioritized based on statistical significance, correlation protein abundance in different morphological features with clinical characteristics, positive staining in the Human Protein Atlas, and limited studies on oral cancers94. IHC was performed for neoplastic islands of 125 cases and stroma of 96 cases. To find out whether the profiles of the biomarker candidates could be seen in saliva, they also performed LC-SRM analyses for 14 cases with no metastatic cancer and 26 cases with metastatic cancer. They found that the expression of CSTB, NDRG1, LTA4H, PGK1, COL6A1 and ITGAV proteins alone or in combination is a good predictor of the disease outcomes and could lead to potential diagnostic assays94.
Chronic kidney disease
In another example of a biomarker study, Good et al. developed a panel of 273 urinary peptides, named CKD273, to study biomarkers of chronic kidney diseases. This panel was developed using a capillary electrophoresis coupled to MS (CE-MS) platform by analyzing a group of 379 health subjects and 230 patients with various biopsy-proven kidney diseases29. CKD273 was developed using a support vector machine model to discriminate between CDK and control groups. This panel was used in a clinical trial to test the performance of the hypertension medicine spironolactone in preventing diabetic nephropathy5. The study followed up 1,775 participants, of which 216 had a high risk of developing diabetic nephropathy, and of these, 209 were included in the trial cohort and were assigned spironolactone (n = 102) or placebo (n = 107). CKD273 was able to predict kidney disease. However, spironolactone failed to prevent progression of the disease155.
Ovarian cancer
Perhaps one of the most successful examples of biomarker development is the OVA1 panel for ovarian cancer. OVA1 panel is composed of CA125, prealbumin, apolipoprotein A1, β2-microglobulin and transferrin, with the last four of them being discovered by surface-enhanced laser desorption ionization (SELDI)-time of flight (TOF) MS13,71,72. In SELDI-TOF, samples are deposited on top of an affinity matrix that binds to limited numbers of proteins based on their physical–chemical properties, reducing the complexity of the samples. Matrices of different properties can be used to bind to different panels of proteins156. Zhang et al. analyzed 57 samples from patients with ovarian cancer paired against 59 healthy controls from two different centers that were divided into two different sets for discovery and cross-validation. Candidate biomarkers were validated against two independent sets with 137 ovarian cancer, 166 benign tumor and 63 healthy control samples. These finding were further validated by immunoassays of another independent set containing 41 ovarian cancer, 20 breast cancer, 20 colon cancer, 20 prostate cancer and 41 healthy control samples71. We should note that, despite the initial promising reports for the discovery and validation of biomarkers, SELDI-TOF was not robust enough for clinical use, and immunological assays were used for biomarker qualification. This is due to the complexity of the instrument, on which small changes in settings can have major impacts on its performance. The time required to perform the measurements is also an important factor as the instrument calibration and detector can drift over time. This is not an issue for ELISA, as whole plates can be read in seconds to a few minutes.
The final assay was tested in the clinic and approved by the Food and Drug Administration (FDA) for clinical use157. However, OVA1 has limited application since it has suboptimal performance for screening patients for ovarian cancer. OVA1 is only used to predict the malignancy of the disease158.
Concluding remarks
There is an urgent need for diagnostics that can be applied to a variety of diseases and conditions. In certain scenarios, including the current coronavirus disease 2019 pandemic, precise tests are needed to diagnose and predict disease outcome. However, biomarker development is a complex task with several phases and multiple failure points. To date, many published biomarker studies are not conclusive or not reproducible because of the failure to consider important factors during project planning and execution. A systematic review of solid tumor biomarkers showed that the low number of samples and lack of proper validation of biomarkers are some of the major challenges of the field159. This highlights that better planning, scientific rigor and QCs are necessary to develop biomarkers that can diagnose or predict the outcome of disease with high accuracy, sensitivity and specificity. Detailed SOPs and consistency during experiments are key elements to ensure reproducibility.
Advances in MS instrumentation will also have a major impact in the field in the near future. Challenges for analyzing an adequate number of samples are the low throughput and high cost of data collection. Typically, a LC-MS/MS run takes 1–2 h to be acquired. However, sample multiplexing with isobaric tags, faster chromatography and additional separation techniques, such as ion mobility spectrometry, have potential to drastically increase the speed and reduce the cost of analysis160,161,162. Therefore, they will have an important role in enabling the analysis of adequate numbers of samples for biomarker development. Technology improvements along with standardized guidelines, such as the one provided by this tutorial, will contribute to the identification of biomarkers that are biologically meaningful and useful in the clinic.
Data availability
All the data discussed in this review are associated with the supporting primary research papers.
References
Rappaport, N. et al. MalaCards: an amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Res. 45, D877–D887 (2017).
Yi, L., Swensen, A. C. & Qian, W. J. Serum biomarkers for diagnosis and prediction of type 1 diabetes. Transl. Res. 201, 13–25 (2018).
Sims, E. K. et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.abc8980 (2021).
Sands, B. E. Biomarkers of inflammation in inflammatory bowel disease. Gastroenterology 149, 1275–1285 e1272 (2015).
Lindhardt, M. et al. Proteomic prediction and Renin angiotensin aldosterone system Inhibition prevention Of early diabetic nephRopathy in TYpe 2 diabetic patients with normoalbuminuria (PRIORITY): essential study design and rationale of a randomised clinical multicentre trial. BMJ Open 6, e010310 (2016).
McShane, L. M. In pursuit of greater reproducibility and credibility of early clinical biomarker research. Clin. Transl. Sci. 10, 58–60 (2017).
Scherer, A. Reproducibility in biomarker research and clinical development: a global challenge. Biomark. Med. 11, 309–312 (2017).
Maes, E., Cho, W. C. & Baggerman, G. Translating clinical proteomics: the importance of study design. Expert Rev. Proteom. 12, 217–219 (2015).
Mischak, H. et al. Implementation of proteomic biomarkers: making it work. Eur. J. Clin. Invest. 42, 1027–1036 (2012).
Frantzi, M., Bhat, A. & Latosinska, A. Clinical proteomic biomarkers: relevant issues on study design & technical considerations in biomarker development. Clin. Transl. Med. 3, 7 (2014).
He, T. Implementation of proteomics in clinical trials. Proteom. Clin. Appl. 13, e1800198 (2019).
Mischak, H. et al. Recommendations for biomarker identification and qualification in clinical proteomics. Sci. Transl. Med. 2, 46ps42 (2010).
Li, D. & Chan, D. W. Proteomic cancer biomarkers from discovery to approval: it’s worth the effort. Expert Rev. Proteom. 11, 135–136 (2014).
Wang, L., McShane, A. J., Castillo, M. J. & Yao, X. in Proteomic and Metabolomic Approaches to Biomarker Discovery 2nd edn (eds Issaq, H. J. & Veenstra, T. D.) 261–288 (Academic Press, 2020).
McNutt, M. Journals unite for reproducibility. Science 346, 679 (2014).
Checklists work to improve science. Nature 556, 273–274 (2018).
Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 533, 452–454 (2016).
European Medicines Agency. Overview of comments received on draft guidance document on qualification of biomarkers. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/overview-comments-received-draft-guidance-document-qualification-biomarkers_en.pdf (2009).
US Food and Drug Administration. Biomarker qualification: evidentiary framework guidance for industry and FDA staff. https://www.fda.gov/media/119271/download (2018).
MacLean, E. et al. A systematic review of biomarkers to detect active tuberculosis. Nat. Microbiol. 4, 748–758 (2019).
Parker, C. E. & Borchers, C. H. Mass spectrometry based biomarker discovery, verification, and validation-quality assurance and control of protein biomarker assays. Mol. Oncol. 8, 840–858 (2014).
Pavlou, M. P. & Diamandis, E. P. in Genomic and Personalized Medicine 2nd edn (eds Ginsburg, G. S. & Huntington, F. W.) 263–271 (Academic Press, 2013).
Kraus, V. B. Biomarkers as drug development tools: discovery, validation, qualification and use. Nat. Rev. Rheumatol. 14, 354–362 (2018).
Masucci, G. V. et al. Validation of biomarkers to predict response to immunotherapy in cancer: volume I—pre-analytical and analytical validation. J. Immunother. Cancer 4, 76 (2016).
Keshishian, H. et al. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 12, 1683–1701 (2017).
Rifai, N., Gillette, M. A. & Carr, S. A. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 (2006).
Shi, T. et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Natl Acad. Sci. USA 109, 15395–15400 (2012).
Ma, M. H. Y. et al. A multi-biomarker disease activity score can predict sustained remission in rheumatoid arthritis. Arthritis Res. Ther. 22, 158 (2020).
Good, D. M. et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol. Cell Proteom. 9, 2424–2437 (2010).
Banerjee, A. & Chaudhury, S. Statistics without tears: populations and samples. Ind. Psychiatry J. 19, 60–65 (2010).
Selvin, S. in Statistical Analysis of Epidemiologic Data. (ed. Selvin, S.) Ch. 4 (Oxford University Press., 2004).
Pearce, N. Analysis of matched case-control studies. BMJ 352, i969 (2016).
Rubin, D. B. Matching to remove bias in observational studies. Biometrics 29, 159–183 (1973).
Mahajan, A. Selection bias: selection of controls as a critical issue in the interpretation of results in a case control study. Indian J. Med. Res. 142, 768 (2015).
Morabia, A. Case-control studies in clinical research: mechanism and prevention of selection bias. Prev. Med. 26, 674–677 (1997).
Sutton-Tyrrell, K. Assessing bias in case-control studies. Proper selection of cases and controls. Stroke 22, 938–942 (1991).
Sheikh, K. Investigation of selection bias using inverse probability weighting. Eur. J. Epidemiol. 22, 349–350 (2007).
Alonso, A. et al. Predictors of follow-up and assessment of selection bias from dropouts using inverse probability weighting in a cohort of university graduates. Eur. J. Epidemiol. 21, 351–358 (2006).
Geneletti, S., Best, N., Toledano, M. B., Elliott, P. & Richardson, S. Uncovering selection bias in case-control studies using Bayesian post-stratification. Stat. Med. 32, 2555–2570 (2013).
VanderWeele, T. J. & Shpitser, I. On the definition of a confounder. Ann. Stat. 41, 196–220 (2013).
Fewell, Z., Davey Smith, G. & Sterne, J. A. The impact of residual and unmeasured confounding in epidemiologic studies: a simulation study. Am. J. Epidemiol. 166, 646–655 (2007).
Polley, M. C. Power estimation in biomarker studies where events are already observed. Clin. Trials 14, 621–628 (2017).
Lalouel, J. M. & Rohrwasser, A. Power and replication in case-control studies. Am. J. Hypertens. 15, 201–205 (2002).
Cai, J. & Zeng, D. Sample size/power calculation for case-cohort studies. Biometrics 60, 1015–1024 (2004).
Jones, S. R., Carley, S. & Harrison, M. An introduction to power and sample size estimation. Emerg. Med. J. 20, 453–458 (2003).
Furberg, C. D. & Friedman, L. M. Approaches to data analyses of clinical trials. Prog. Cardiovasc. Dis. 54, 330–334 (2012).
Levin, Y. The role of statistical power analysis in quantitative proteomics. Proteomics 11, 2565–2567 (2011).
Dicker, L., Lin, X. & Ivanov, A. R. Increased power for the analysis of label-free LC-MS/MS proteomics data by combining spectral counts and peptide peak attributes. Mol. Cell Proteom. 9, 2704–2718 (2010).
Skates, S. J. et al. Statistical design for biospecimen cohort size in proteomics-based biomarker discovery and verification studies. J. Proteome Res. 12, 5383–5394 (2013).
Webb-Robertson, B. M. et al. Statistically driven metabolite and lipid profiling of patients from the undiagnosed diseases network. Anal. Chem. 92, 1796–1803 (2020).
Nakayasu, E. S. et al. Comprehensive proteomics analysis of stressed human islets identifies GDF15 as a target for type 1 diabetes intervention. Cell Metab. 31, 363–374 e366 (2020).
Ocaña, G. J. et al. Analysis of serum Hsp90 as a potential biomarker of β cell autoimmunity in type 1 diabetes. PLoS ONE 14, e0208456 (2019).
Sims, E. K. et al. Elevations in the fasting serum proinsulin-to-C-peptide ratio precede the onset of type 1 diabetes. Diabetes Care 39, 1519–1526 (2016).
Townsend, M. K. et al. Impact of pre-analytic blood sample collection factors on metabolomics. Cancer Epidemiol. Biomark. Prev. 25, 823–829 (2016).
Cemin, R. & Daves, M. Pre-analytic variability in cardiovascular biomarker testing. J. Thorac. Dis. 7, E395–E401 (2015).
Pasic, M. D. et al. Influence of fasting and sample collection time on 38 biochemical markers in healthy children: a CALIPER substudy. Clin. Biochem. 45, 1125–1130 (2012).
Narayanan, S. The preanalytic phase. An important component of laboratory medicine. Am. J. Clin. Pathol. 113, 429–452 (2000).
Stewart, T. et al. Impact of pre-analytical differences on biomarkers in the ADNI and PPMI studies: implications in the era of classifying disease based on biomarkers. J. Alzheimers Dis. 69, 263–276 (2019).
Speake, C. et al. Circulating unmethylated insulin DNA as a biomarker of human beta cell death: a multi-laboratory assay comparison. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgaa008 (2020).
Holst, J. J. & Wewer Albrechtsen, N. J. Methods and guidelines for measurement of glucagon in plasma. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20215416 (2019).
Steiner, C. et al. Applications of mass spectrometry for quantitative protein analysis in formalin-fixed paraffin-embedded tissues. Proteomics 14, 441–451 (2014).
Giusti, L., Angeloni, C. & Lucacchini, A. Update on proteomic studies of formalin-fixed paraffin-embedded tissues. Expert Rev. Proteom. 16, 513–520 (2019).
Piehowski, P. D. et al. Residual tissue repositories as a resource for population-based cancer proteomic studies. Clin. Proteom. 15, 26 (2018).
Thompson, S. M. et al. Impact of pre-analytical factors on the proteomic analysis of formalin-fixed paraffin-embedded tissue. Proteom. Clin. Appl. 7, 241–251 (2013).
Pellis, L. et al. Plasma metabolomics and proteomics profiling after a postprandial challenge reveal subtle diet effects on human metabolic status. Metabolomics 8, 347–359 (2012).
Johansen, P., Andersen, J. D., Børsting, C. & Morling, N. Evaluation of the iPLEX® Sample ID Plus Panel designed for the Sequenom MassARRAY® system. A SNP typing assay developed for human identification and sample tracking based on the SNPforID panel. Forensic Sci. Int. Genet. 7, 482–487 (2013).
Hoofnagle, A. N. et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry-based assays. Clin. Chem. 62, 48–69 (2016).
Sims, E. K. et al. Proinsulin secretion is a persistent feature of type 1 diabetes. Diabetes Care 42, 258–264 (2019).
Schulz, K. F. & Grimes, D. A. Blinding in randomised trials: hiding who got what. Lancet 359, 696–700 (2002).
Karanicolas, P. J., Farrokhyar, F. & Bhandari, M. Practical tips for surgical research: blinding: who, what, when, why, how? Can. J. Surg. 53, 345–348 (2010).
Zhang, Z. et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 64, 5882–5890 (2004).
Zhang, Z. & Chan, D. W. The road from discovery to clinical diagnostics: lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol. Biomark. Prev. 19, 2995–2999 (2010).
Anderson, N. L. & Anderson, N. G. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell Proteom. 1, 845–867 (2002).
Liu, H., Sadygov, R. G. & Yates, J. R. 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 (2004).
Qian, W. J. et al. Enhanced detection of low abundance human plasma proteins using a tandem IgY12-SuperMix immunoaffinity separation strategy. Mol. Cell Proteom. 7, 1963–1973 (2008).
Liu, T. et al. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol. Cell Proteom. 5, 2167–2174 (2006).
Yadav, A. K. et al. A systematic analysis of eluted fraction of plasma post immunoaffinity depletion: implications in biomarker discovery. PLoS ONE 6, e24442 (2011).
Garay-Baquero, D. J. et al. Comprehensive plasma proteomic profiling reveals biomarkers for active tuberculosis. JCI Insight https://doi.org/10.1172/jci.insight.137427 (2020).
Piehowski, P. D. et al. Sources of technical variability in quantitative LC-MS proteomics: human brain tissue sample analysis. J. Proteome Res. 12, 2128–2137 (2013).
Wisniewski, J. R., Ostasiewicz, P. & Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 10, 3040–3049 (2011).
Quesada-Calvo, F. et al. Comparison of two FFPE preparation methods using label-free shotgun proteomics: application to tissues of diverticulitis patients. J. Proteom. 112, 250–261 (2015).
Kawashima, Y., Kodera, Y., Singh, A., Matsumoto, M. & Matsumoto, H. Efficient extraction of proteins from formalin-fixed paraffin-embedded tissues requires higher concentration of tris(hydroxymethyl)aminomethane. Clin. Proteom. 11, 4 (2014).
Kulevich, S. E., Frey, B. L., Kreitinger, G. & Smith, L. M. Alkylating tryptic peptides to enhance electrospray ionization mass spectrometry analysis. Anal. Chem. 82, 10135–10142 (2010).
Walmsley, S. J. et al. Comprehensive analysis of protein digestion using six trypsins reveals the origin of trypsin as a significant source of variability in proteomics. J. Proteome Res. 12, 5666–5680 (2013).
Herraiz, T. & Casal, V. Evaluation of solid-phase extraction procedures in peptide analysis. J. Chromatogr. A 708, 209–221 (1995).
Muntel, J. et al. Comparison of protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J. Proteome Res. 18, 1340–1351 (2019).
Ow, S. Y. et al. iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J. Proteome Res. 8, 5347–5355 (2009).
Liu, Y. et al. Quantitative variability of 342 plasma proteins in a human twin population. Mol. Syst. Biol. 11, 786 (2015).
Geyer, P. E. et al. Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Mol. Syst. Biol. 12, 901 (2016).
Bekker-Jensen, D. B. et al. A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients. Mol. Cell Proteom. 19, 716–729 (2020).
Xuan, Y. et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nat. Commun. 11, 5248 (2020).
Shen, Y. et al. Discovery of potential plasma biomarkers for tuberculosis in HIV-infected patients by data-independent acquisition-based quantitative proteomics. Infect. Drug Resist. 13, 1185–1196 (2020).
Fang, X. et al. Urinary proteomics of Henoch-Schonlein purpura nephritis in children using liquid chromatography-tandem mass spectrometry. Clin. Proteom. 17, 10 (2020).
Carnielli, C. M. et al. Combining discovery and targeted proteomics reveals a prognostic signature in oral cancer. Nat. Commun. 9, 3598 (2018).
Bekker-Jensen, D. B. et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 4, 587–599 e584 (2017).
Ow, S. Y., Salim, M., Noirel, J., Evans, C. & Wright, P. C. Minimising iTRAQ ratio compression through understanding LC-MS elution dependence and high-resolution HILIC fractionation. Proteomics 11, 2341–2346 (2011).
Manadas, B., Mendes, V. M., English, J. & Dunn, M. J. Peptide fractionation in proteomics approaches. Expert Rev. Proteom. 7, 655–663 (2010).
Schoenmakers, P. J., van Molle, S., Hayes, C. M. G. & Uunk, L. G. M. Effects of pH in reversed-phase liquid chromatography. Anal. Chim. Acta 250, 1–19 (1991).
Amidan, B. G. et al. Signatures for mass spectrometry data quality. J. Proteome Res. 13, 2215–2222 (2014).
Zhang, T. et al. Block design with common reference samples enables robust large-scale label-free quantitative proteome profiling. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00310 (2020).
Burger, B., Vaudel, M. & Barsnes, H. Importance of block randomization when designing proteomics experiments. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00536 (2020).
Stanfill, B. A. et al. Quality control analysis in real-time (QC-ART): a tool for real-time quality control assessment of mass spectrometry-based proteomics data. Mol. Cell Proteom. 17, 1824–1836 (2018).
Matzke, M. M. et al. Improved quality control processing of peptide-centric LC-MS proteomics data. Bioinformatics 27, 2866–2872 (2011).
Bittremieux, W., Valkenborg, D., Martens, L. & Laukens, K. Computational quality control tools for mass spectrometry proteomics. Proteomics https://doi.org/10.1002/pmic.201600159 (2017).
Devabhaktuni, A. et al. TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat. Biotechnol. 37, 469–479 (2019).
Cox, J. et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011).
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277 (2014).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Gan, N. et al. Regulation of phosphoribosyl ubiquitination by a calmodulin-dependent glutamylase. Nature 572, 387–391 (2019).
Callister, S. J. et al. Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res. 5, 277–286 (2006).
Kultima, K. et al. Development and evaluation of normalization methods for label-free relative quantification of endogenous peptides. Mol. Cell Proteom. 8, 2285–2295 (2009).
Webb-Robertson, B. J., Matzke, M. M., Jacobs, J. M., Pounds, J. G. & Waters, K. M. A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics 11, 4736–4741 (2011).
Valikangas, T., Suomi, T. & Elo, L. L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief. Bioinform. 19, 1–11 (2018).
Karpievitch, Y. V., Dabney, A. R. & Smith, R. D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics 13, S5 (2012).
Liebal, U. W., Phan, A. N. T., Sudhakar, M., Raman, K. & Blank, L. M. Machine learning applications for mass spectrometry-based metabolomics. Metabolites https://doi.org/10.3390/metabo10060243 (2020).
Kim, M., Rai, N., Zorraquino, V. & Tagkopoulos, I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 7, 13090 (2016).
Sedgwick, P. Multiple hypothesis testing and Bonferroni’s correction. BMJ 349, g6284 (2014).
Artigaud, S., Gauthier, O. & Pichereau, V. Identifying differentially expressed proteins in two-dimensional electrophoresis experiments: inputs from transcriptomics statistical tools. Bioinformatics 29, 2729–2734 (2013).
Strimmer, K. A unified approach to false discovery rate estimation. BMC Bioinformatics 9, 303 (2008).
Frohnert, B. I. et al. Predictive modeling of type 1 diabetes stages using disparate data sources. Diabetes 69, 238–248 (2020).
Sonsare, P. M. & Gunavathi, C. Investigation of machine learning techniques on proteomics: a comprehensive survey. Prog. Biophys. Mol. Biol. 149, 54–69 (2019).
Palivec, V. [Minutiae, the first Czech medical prints]. Cas. Lek. Cesk 128, 1530 (1989).
Colby, S. M., McClure, R. S., Overall, C. C., Renslow, R. S. & McDermott, J. E. Improving network inference algorithms using resampling methods. BMC Bioinformatics 19, 376 (2018).
Schiess, R., Wollscheid, B. & Aebersold, R. Targeted proteomic strategy for clinical biomarker discovery. Mol. Oncol. 3, 33–44 (2009).
Surinova, S. et al. On the development of plasma protein biomarkers. J. Proteome Res. 10, 5–16 (2011).
Burgess, M. W., Keshishian, H., Mani, D. R., Gillette, M. A. & Carr, S. A. Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol. Cell Proteom. 13, 1137–1149 (2014).
Kennedy, J. J. et al. Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins. Nat. Methods 11, 149–155 (2014).
Kim, Y. et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nat. Commun. 7, 11906 (2016).
Paulovich, A. G., Whiteaker, J. R., Hoofnagle, A. N. & Wang, P. The interface between biomarker discovery and clinical validation: the tar pit of the protein biomarker pipeline. Proteom. Clin. Appl. 2, 1386–1402 (2008).
Kawahara, R. et al. Integrative analysis to select cancer candidate biomarkers to targeted validation. Oncotarget 6, 43635–43652 (2015).
Toth, R. et al. Random forest-based modelling to detect biomarkers for prostate cancer progression. Clin. Epigenetics 11, 148 (2019).
Olivier, M., Asmis, R., Hawkins, G. A., Howard, T. D. & Cox, L. A. the need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20194781 (2019).
Lange, V., Picotti, P., Domon, B. & Aebersold, R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222 (2008).
Tarasova, I. A., Masselon, C. D., Gorshkov, A. V. & Gorshkov, M. V. Predictive chromatography of peptides and proteins as a complementary tool for proteomics. Analyst 141, 4816–4832 (2016).
Rost, H., Malmstrom, L. & Aebersold, R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol. Cell Proteom. 11, 540–549 (2012).
Mueller, L. K., Baumruck, A. C., Zhdanova, H. & Tietze, A. A. Challenges and perspectives in chemical synthesis of highly hydrophobic peptides. Front. Bioeng. Biotechnol. 8, 162 (2020).
Wu, C. et al. Expediting SRM assay development for large-scale targeted proteomics experiments. J. Proteome Res. 13, 4479–4487 (2014).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Pino, L. K. et al. Matrix-matched calibration curves for assessing analytical figures of merit in quantitative proteomics. J. Proteome Res. 19, 1147–1153 (2020).
Whiteaker, J. R. et al. CPTAC Assay Portal: a repository of targeted proteomic assays. Nat. Methods 11, 703–704 (2014).
Yu, L. et al. Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann. Neurol. 84, 78–88 (2018).
Whiteaker, J. R. et al. Peptide immunoaffinity enrichment with targeted mass spectrometry: application to quantification of ATM kinase phospho-signaling. Methods Mol. Biol. 1599, 197–213 (2017).
Zhu, Y. et al. Immunoaffinity microflow liquid chromatography/tandem mass spectrometry for the quantitation of PD1 and PD-L1 in human tumor tissues. Rapid Commun. Mass Spectrom. 34, e8896 (2020).
Schneck, N. A., Phinney, K. W., Lee, S. B. & Lowenthal, M. S. Quantification of cardiac troponin I in human plasma by immunoaffinity enrichment and targeted mass spectrometry. Anal. Bioanal. Chem. 410, 2805–2813 (2018).
Sall, A. et al. Advancing the immunoaffinity platform AFFIRM to targeted measurements of proteins in serum in the pg/ml range. PLoS ONE 13, e0189116 (2018).
Jung, S. et al. Quantification of ATP7B protein in dried blood spots by peptide immuno-SRM as a potential screen for Wilson’s disease. J. Proteome Res. 16, 862–871 (2017).
Schoenherr, R. M. et al. Multiplexed quantification of estrogen receptor and HER2/Neu in tissue and cell lysates by peptide immunoaffinity enrichment mass spectrometry. Proteomics 12, 1253–1260 (2012).
Gibbons, B. C. et al. Rapidly assessing the quality of targeted proteomics experiments through monitoring stable-isotope labeled standards. J. Proteome Res. 18, 694–699 (2019).
Carr, S. A. et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell Proteom. 13, 907–917 (2014).
Grant, R. P. & Hoofnagle, A. N. From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 60, 941–944 (2014).
Chen, Z. et al. Quantitative insulin analysis using liquid chromatography-tandem mass spectrometry in a high-throughput clinical laboratory. Clin. Chem. 59, 1349–1356 (2013).
Zhang, Q. et al. Serum proteomics reveals systemic dysregulation of innate immunity in type 1 diabetes. J. Exp. Med. 210, 191–203 (2013).
Almangush, A. et al. A simple novel prognostic model for early stage oral tongue cancer. Int. J. Oral. Maxillofac. Surg. 44, 143–150 (2015).
Tofte, N. et al. Early detection of diabetic kidney disease by urinary proteomics and subsequent intervention with spironolactone to delay progression (PRIORITY): a prospective observational study and embedded randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 8, 301–312 (2020).
Issaq, H. J., Veenstra, T. D., Conrads, T. P. & Felschow, D. The SELDI-TOF MS approach to proteomics: protein profiling and biomarker identification. Biochem. Biophys. Res. Commun. 292, 587–592 (2002).
Fung, E. T. A recipe for proteomics diagnostic test development: the OVA1 test, from biomarker discovery to FDA clearance. Clin. Chem. 56, 327–329 (2010).
Carvalho, V. P. et al. The contribution and perspectives of proteomics to uncover ovarian cancer tumor markers. Transl. Res. 206, 71–90 (2019).
Belczacka, I. et al. Proteomics biomarkers for solid tumors: current status and future prospects. Mass Spectrom. Rev. 38, 49–78 (2019).
Ma, J. & Kilby, G. W. Sensitive, rapid, robust, and reproducible workflow for host cell protein profiling in biopharmaceutical process development. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.0c00252 (2020).
Couvillion, S. P. et al. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst 144, 794–807 (2019).
Li, J. et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 17, 399–404 (2020).
Beausoleil, S. A., Villen, J., Gerber, S. A., Rush, J. & Gygi, S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006).
Acknowledgements
The authors thank N. Johnson for his help in designing figures used in this publication. This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants UC4 DK104166 (to C.E.M. and T.O.M.), U01 DK127786 (to C.E.M., B.J.M.W.R and T.O.M.), U01 DK124020 (to W.J.Q), R01 DK032493 (to M.R.) and P30DK097512 (to C.E.M), R01 DK093954 (to C.E.M) and R21 DK119800-01A1 (to C.E.M). M.R., E.S.N., B.J.M.W.R. and T.O.M. were also supported by the Helmsley Trust grant G-1901-03687. C.E.M was also supported by VA Merit Award I01BX001733, JDRF 2-SRA-2018-493-A-B and gifts from the Sigma Beta Sorority, the Ball Brothers Foundation, and the George and Frances Ball Foundation. The TEDDY Study is funded by U01 DK63829, U01 DK63861, U01 DK63821, U01 DK63865, U01 DK63863, U01 DK63836, U01 DK63790, UC4 DK63829, UC4 DK63861, UC4 DK63821, UC4 DK63865, UC4 DK63863, UC4 DK63836, UC4 DK95300, UC4 DK100238, UC4 DK106955, UC4 DK112243, UC4 DK117483, and Contract No. HHSN267200700014C from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Environmental Health Sciences (NIEHS), Centers for Disease Control and Prevention (CDC), and JDRF. TEDDY is supported in part by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida (UL1 TR000064) and the University of Colorado (UL1 TR002535). Work was performed in the Environmental Molecular Sciences Laboratory, a US Department of Energy (DOE) national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, WA. Battelle operates PNNL for the DOE under contract DE-AC05-76RLO01830.
Author information
Authors and Affiliations
Contributions
E.S.N. wrote the abstract, introduction and concluding remarks, contributed to the data analysis section and edited the manuscript; A.M.D.S., J.P.K., M.R. and B.I.F. wrote the sections on subject selection, power calculation and considerations for sample handling. C.E.M. wrote the section on specimen collection, storage and tracking; M.G., P.D.P. and A.S. wrote the sample preparation sections of both discovery and validation phases; D.O. wrote the section on data collection for the discovery phase; C.A. wrote the section on data quality control; B.J.W.R. contributed to the power analysis section and wrote the data analysis section; Y.G., P.D.P., T.F. and W.J.Q wrote about the different sections of the validation phase; T.O.M. wrote the phases of biomarker development. All the authors read, provided inputs and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Protocols thanks Bing Zhang and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this review
Zhang, Q. et al. J. Exp. Med. 210, 191–203 (2013): https://doi.org/10.1084/jem.20111843
Carnielli, C. M. et al. Nat. Commun. 9, 3598 (2018): https://doi.org/10.1038/s41467-018-05696-2
Tofte, N. et al. Lancet Diabetes Endocrinol. 8, 301–312 (2020): https://doi.org/10.1016/S2213-8587(20)30026-7
Zhang, Z. et al. Cancer Res. 64, 5882–5890 (2004): https://doi.org/10.1158/0008-5472.CAN-04-0746
Rights and permissions
About this article

Cite this article
Nakayasu, E.S., Gritsenko, M., Piehowski, P.D. et al. Tutorial: best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat Protoc 16, 3737–3760 (2021). https://doi.org/10.1038/s41596-021-00566-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00566-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
