
Abstract
The global pandemic of coronavirus disease 2019 (COVID-19) highlights the shortcomings of the current testing paradigm for viral disease diagnostics. Here, we report a stepwise protocol for an RNA-extraction-free nano-amplified colorimetric test for rapid and naked-eye molecular diagnosis of COVID-19. The test employs a unique dual-prong approach that integrates nucleic acid (NA) amplification and plasmonic sensing for point-of-care detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), with a sample-to-assay response time of <1 h. The RNA-extraction-free nano-amplified colorimetric test utilizes plasmonic gold nanoparticles capped with antisense oligonucleotides (ASOs) as a colorimetric reporter to detect the amplified nucleic acid from the COVID-19 causative virus, SARS-CoV-2. The ASOs are specific for the SARS-CoV-2 N-gene, and binding of the ASOs to their target sequence results in the aggregation of the plasmonic gold nanoparticles. This highly specific agglomeration step leads to a change in the plasmonic response of the nanoparticles. Furthermore, when tested using clinical samples, the accuracy, sensitivity and specificity of the test were found to be >98.4%, >96.6% and 100%, respectively, with a detection limit of 10 copies/μL. The test can easily be adapted to diagnose other viral infections with a simple modification of the ASOs and primer sequences. It also provides a low-cost, rapid approach requiring minimal instrumentation that can be used as a screening tool for the diagnosis of COVID-19 at point-of-care settings in resource-poor situations. The colorimetric readout of the test can even be monitored using a handheld optical reader to obtain a quantitative response. Therefore, we anticipate that this protocol will be widely useful for the development of biosensors for the molecular diagnostics of COVID-19 and other infectious diseases.
Similar content being viewed by others


Introduction
Being a blueprint of our genetic data, nucleic acids (NA) underly the continuity of life1. Though the genetic material of almost every living organism is composed of DNA or RNA, variations exist in their genome sequences. This creates inherent differences among races and ethnic populations, as well as between healthy individuals and individuals vulnerable to diseases2. Therefore, the rapid identification of specific NA has enormous potential in disease diagnosis and management3. One of the most common applications of NA detection is molecular-based diagnostic tests or NA testing for the diagnosis of various infectious diseases caused by different microbes and pathogens4,5,6,7.
NA testing (NAT) or NA amplification testing (NAAT) is a process that involves amplification and identification of NA for diagnosis and/or guidance of therapy8. Commercially available NA detection methodologies typically involve amplification of NA extracted from the biofluids collected from patients under observation9. Globally, infectious diseases have an enormous impact on the community from a social as well as economic standpoint10,11. For example, more than 100 million people are infected worldwide with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that is responsible for the coronavirus disease 2019 (COVID-19) pandemic, leading to >2 million deaths as of February 2021 and substantial socioeconomic disruption. The effective and large-scale diagnostic methods needed for the selective and sensitive detection of the rapid dissemination of the virus in the community are a crucial aspect of the management of the COVID-19 pandemic12,13. Current standard practice for detecting an active COVID-19 infection uses reverse-transcriptase real-time polymerase chain reaction (RT-PCR)14, which requires labor-intensive, laboratory-based protocols for viral RNA isolation, lysis and removal of inhibiting materials. While PCR remains the proven gold standard for clinical diagnostics, these restrictions become major hurdles for situations with resource scarcity. Therefore, there is an immediate need to develop instrument-free approaches that are low-cost and rapid and can be used as a screening tool for the diagnosis of COVID-19 infection at point-of-care (POC)14. Rapid, low-cost and user-friendly molecular diagnostic methods are important for combating outbreaks of infectious diseases15,16. Especially during the current pandemic of COVID-19, it is critical to expand the testing capacity beyond laboratory settings. There is an immediate need to develop global testing capability up to 100-fold over existing conventional approaches. In recognition of this unmet need, the World Health Organization has established the ASSURED criteria (Affordable, Sensitive, Specific, User-friendly, Rapid and robust, Equipment-free and Deliverable), to which these tests should adhere if they are to be effective in resource-limited environments17.
Based on these requirements, new diagnostic approaches for COVID-19 and other infectious diseases are in high demand. Loop-mediated isothermal amplification (LAMP) technology has been widely used for various NAAT approaches18,19,20. LAMP is similar to the traditional PCR tests, except that NA amplification happens at a set temperature20,21,22,23. Therefore, this innovative procedure does not require specific equipment, such as a thermal cycler, which makes it simpler, more user-friendly and more cost-effective than PCR-based approaches, particularly in the POC diagnostic scenario. Other advantages of LAMP include the accommodation of a wide pH and temperature range, ability to work with nonprocessed samples and flexibility of readout methods without compromising the sensitivity compared with the PCR tests. Although LAMP-based testing approaches such as colorimetric reverse-transcriptase LAMP (RT-LAMP)23,24 provide sensitive and accurate results, the method still suffers from a high incidence of false-positive results due to the presence of spurious amplification byproducts25. There are also challenges in using LAMP for multiplex assays in a single sample and for the quantitation of target genetic material.
Nanoparticles and nanocomposites play a crucial role in sensing5,6,26,27,28,29,30,31,32,33,34,35, imaging36,37,38,39 and therapy40,41,42,43,44,45,46, and recently, antisense oligonucleotide (ASO)-capped nanoparticles have been utilized for rapid, sensitive, easy-to-use and portable detection of NA6,7. ASOs represent a highly promising class of single-stranded DNA or RNA that are complementary to a given target sequence47. Their rational design and affordability and the progress in human genome sequencing have all contributed to the use of ASOs either as therapeutic agents or as key elements to study the genetic function48. For example, these short fragments can target both DNA and RNA, (including pre‐mRNA, mRNA and ncRNA) to inhibit gene expression and can thus induce a blockade in the transfer of genetic information from DNA to protein47. ASOs could be designed to be unique for each molecular and genetic change and be applied to restore a normal physiological condition47. However, while ASOs are widely used both in the laboratory and clinic as therapeutic agents, their applicability as molecular probes for detecting specific NA sequences remains relatively unexplored49.
Development of the protocol
To overcome the aforementioned limitations of the currently available COVID-19 tests, we developed a unique nanotechnology-based molecular sensing approach that can be adapted to accurately detect a target NA6,7. Our ‘nano-amplified colorimetric test’ (NACT) uses ASO-capped plasmonic nanoparticles for selective, and ‘naked-eye’ detection of SARS-CoV-2 (ref. 6). We have optimized the described protocol in our previously published reports.6,7 In these two reports, we designed ASO sequences targeting N-gene segment of SARS-CoV-2 RNA. We also suitably functionalized the selected ASOs to conjugate them with citrate-stabilized gold nanoparticles. The visual change in the plasmonic response of the nanoparticles upon their selective aggregation in the presence of the target SARS-CoV-2 RNA was also described in our previously published report6. The aggregation of the ASO-capped plasmonic gold nanoparticles (AuNPs) using COVID-19 clinical samples was also confirmed in our previous report7. This protocol is an extension of our previous work5,6,7 with modifications made to achieve higher sensitivity and specificity for SARS-CoV-2 detection. To address the low viral copy number present in the clinical samples and to enable the protocol to be generalizable for other relevant viruses, we have introduced a NA amplification step based on our previous report5 of the plasmonic detection methodology reported in refs. 6,7. During the study, we also realized that extraction of RNA is not a requirement for our protocol; rather, the technology can be applied directly to the clinical samples without a prior RNA extraction step and with only the addition of suitable lysis buffer. Hence, this protocol shows data relating specifically to development of the protocol in terms of validation, benchmarking and troubleshooting.
Briefly, in refs. 6,7 we targeted the N-gene sequence of SARS-CoV-2 isolate 2019-nCoV/USA-WA1-A12/2020 and designed a set of ASOs. We have selected potential ASOs on the basis of their target binding energy and disruption energy. Pairs of ASOs are chosen that bind to closely spaced regions in the target sequences6,7. Each of the ASOs in a pair was functionalized with a thiol moiety either at the 5′ or 3′ end and then used to cap AuNPs. This functionalization approach was followed so that the thiol groups could come close to each other when the two ASOs are bound to the target sequence6,7. Thus, as the thiol groups are conjugated to gold nanoparticles, they cause the gold nanoparticles to come close to each other only in the presence of the target sequence (the N-gene sequence of SARS-CoV-2) leading to agglomeration among nanoparticles that can be observed by the change in the surface plasmon band (Supplementary Fig. 1).
Although this was a successful demonstration of the use of ASO combined with plasmonic nanoparticles for selection and detection of the SARS-CoV-2 genetic sequence requiring minimal equipment, the assay still had a notable limitation6. The assay had a relatively high detection limit, which resulted in erroneous detection of samples as false negatives6. To address this limitation, we introduced an isothermal NA amplification step5 to improve the limit of detection of the assay so that it could detect samples with much lower viral copy numbers. Accordingly, we designed specific primer sets (inner, outer and loop) for the targeted region of the SARS-CoV-2 N-gene (see Experimental design)5. We have further investigated the versatility of our approach using nasal and oropharyngeal swabs collected from confirmed positive and negative COVID-19 samples7.
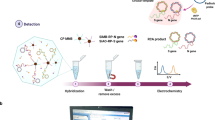
In refs. 6,7 we demonstrated that our developed protocol for COVID-19 diagnosis is a robust approach that can diagnose COVID-19 and differentiate positive samples from negatives as well as cross-reactive samples such as other coronaviruses. Although the developed technology showed an excellent result when tested with extracted RNA from clinical samples7 and artificial saliva spiked with purified RNAs (M.A., P.M., K.D. and D.P., unpublished results), clinical samples typically contain other components, many of which might interfere with the sensing protocol when tested without RNA extraction6,7. Therefore, optimizing the test protocol for using clinical samples directly, without RNA extraction, was necessary to avoid any interference and to create an RNA-extraction-free protocol for SARS-CoV-2 detection. RNA-extraction-free methodology has received a great deal of attention lately, as it enables individuals with limited scientific expertise to conduct the test and would make it easier to expand the test applicability to limited resource areas and mass gatherings such as airports and schools50. The diagnostic results using our protocol can be visualized within 40 min by the naked eye with no sample preprocessing required, making the RNA extraction step optional for the test (M.A., P.M., K.D. and D.P., unpublished results). The amplified sample volume can be as low as 10 μl (ref. 5). The limit of detection of the test was found to be 10 viral copies per μl (M.A., P.M., K.D. and D.P., unpublished results). Furthermore, no change in color was observed in negative samples with a specificity of 100% (M.A., P.M., K.D. and D.P., unpublished results, Supplementary Table 1). The Procedure includes NACT kit design, reagent preparation, design of ASOs, design of nucleic acid amplification (NAA) primers, and design and preparation of nanomaterials for the clinical detection of SARS-CoV-2 genetic material as shown in Fig. 1. The experimental design and data interpretation are also discussed in detail.

First, the NAA primers and ASOs are designed and purchased from commercial vendors (Experimental design). The AuNPs are synthesized and capped with the designed ASOs (Steps 1–7). Samples (nasal swab/ saliva) are collected and either lysed and used directly, or, optionally, extraction of RNA can be performed (Steps 8–9). The NA is then isothermally amplified (Steps 10–15) and detected using the Au-ASOTEST solution containing ASO-capped Au nanoparticles (Steps 16–17). Detection can be performed simply by observing the color change by the naked eye or by recording the absorbance signal using a portable reader (Picoexplorer) or a plate reader.
Advantages and limitations
Our NAA-based NACT has several advantages over the alternative currently available molecular techniques (discussed further below). There are five main characteristics that set our test apart from other approaches available for COVID-19 detection: (i) it does not need prior RNA extraction; (ii) it does not demand the use of advanced equipment (e.g., centrifuge, thermocycler, etc.); (iii) it does not use conventional pH-sensitive dyes, instead using specific ASOs for targeting a selected region of N gene conjugated to plasmonic AuNPs to indicate a color change, permitting a simple colorimetric readout and enabling label-free sequence-specific detection (i.e., no fluorescence labeling is required such as in PCR); (iv) it uses low-cost (Supplementary Table 2) and easily accessible reagents that can be rapidly manufactured in bulk for commercialization (i.e., for the future commercialization of NACT kit); and (v) it has a short turnaround time (Supplementary Table 3).
The quick turnaround of the results is an attractive feature of the developed NACT kit. The NAA reaction can be carried out within 30 min, and part of this solution is transferred to the Au-ASO colorimetric reaction as part of a two-step procedure that can detect the SARS-CoV-2 RNA target. Another advantage of the colorimetric NACT kit over other developed platforms such as RT-PCR and clustered regularly interspaced short palindromic repeats (CRISPR)-based tests is the low cost of the kit components51,52. For example, AuNPs can be produced at a large scale, and the ASOs are short single-stranded DNA sequences that are relatively economical to synthesize. The rest of the kit reagents and enzymatic components can also be purchased in bulk. Our NACT test can be designed and synthesized in a very short time for as little as $0.15 per test (Supplementary Table 2). Moreover, this NACT method enables label-free sequence-specific detection, with a simple colorimetric readout, and the avoidance of NA extraction. The isolation and purification of NA from clinical samples is generally based on the use of expensive commercial kits containing several steps and specialized laboratory equipment such as centrifuges. Our NACT approach utilizes only lysis buffer to rupture the viral membrane present in clinical samples and uses the lysis reaction directly for the amplification reaction. This enables the rapid detection of SARS-CoV-2 infection at a POC level.
Despite several advantages of this approach over other methodologies, there are limitations that require closer attention. The design of ASOs and NAA primers involves a close understanding of molecular biology and careful investigation of the parameters associated with each candidate to determine the optimal ASO and primer combination. Furthermore, because the NACT kit includes an isothermal NAA phase, there is a high chance that contamination will be observed in subsequent reactions, leading to false-positive results if the NAA procedure has not been carried out in a suitably clean environment23,53,54. Another possible drawback of the NACT assay is that it depends on the user’s viewpoint and color perception to decide on the diagnosis of the sample (i.e., positive or negative); however, this can be addressed by introducing a plasmonic optical readout. Also, this NACT method accurately detects the target genetic material in two steps and is not recommended for accurate gene expression profiling.
Applications of the method
In the midst of this latest once-in-a-century global pandemic, the potential applicability of our RNA-extraction-free approach can provide widespread availability of a COVID-19 diagnostic test, especially to non-laboratory environments such as airports, grocery stores, hospitals, emergency treatment facilities and remote research centers50. This newly developed protocol can be used as a key approach in rapid and large-scale diagnosis of COVID-19, as well as to build a POC platform to diagnose other infectious diseases caused by various microbial and viral pathogens by changing the configuration of NAA primers and corresponding ASOs. We have successfully used this technique to rapidly identify SARS-CoV-2 RNA from a wide variety of sample types, including oropharyngeal, nasal swab samples6,7 and artificial saliva spiked with purified SARS-CoV-2 RNA (M.A., P.M., K.D. and D.P., unpublished results). Furthermore, this protocol has widespread applications, and is just not limited to SARS-CoV-2. The applicability of this protocol can be further expanded to cover other infectious diseases caused by other viruses or bacteria55,56 by simply redesigning the ASO sequences to selectively target RNA of the varying biological targets.
Comparison with other methods
While PCR remains the proven gold standard for clinical diagnostics, there is an urgent need for other approaches that are low-cost and rapid and provide diagnosis at the POC level51. Therefore, new solutions and methodologies for NA detection are in high demand. One such method is LAMP57. At present, the commercially available RT-LAMP-based colorimetric technologies for the diagnosis of COVID-19 typically involve isolating RNA, amplifying the desired segment and detecting the isolated genetic material20,21,25. While the commercial colorimetric RT-LAMP COVID-19 tests are excellent in terms of sensitivity, carry‐over contamination is a common drawback in such reactions, which usually leads to false-positive results and decreased selectivity23. Moreover, if the RT-LAMP technique is not performed following good molecular biology practices, carryover contamination can be observed in subsequent reactions, leading to false-positive results. Several research groups have developed different versions of LAMP-based molecular diagnostic tests such as iLACO58, one-pot RT-LAMP59,60, mismatch-tolerant RT-LAMP61, Penn-RAMP62, RT-LAMP + CRISPR-Cas1263, and STOPCovid (SHERLOCK)64. While these tests are remarkable in terms of selectivity and sensitivity, they have their inherit limitations. For example, the Penn-RAMP method combines another isothermal amplification method with the traditional LAMP62. In this case, first, a recombinase polymerase amplification (RPA) process is conducted in the cap of a test tube at 38 °C for ~15–20 min. During this RPA process, an enzyme called recombinase assists the forward outer primer (F3) and backward outer primer (B3) LAMP primers in locating the targeted sequence of the sample. In the next step, the RPA mixture is mixed with conventional LAMP reagents to detect the presence of NA. Although the technique is excellent in terms of sensitivity, it still has certain drawbacks such as RPA undergoing asynchronous amplification with the potential to saturate, which can prevent accurate quantitation. Moreover, relying on two isothermal amplification approaches makes the system complicated and increases the necessary reagents and the test cost. In another study, RT-LAMP was integrated with CRISPR-Cas12 for the detection of COVID-19. This novel CRISPR-enabled diagnostic tool, called SARS-CoV-2 DNA endonuclease-targeted CRISPR trans reporter (DETECTR)63, uses the enzyme Cas12 after RT-LAMP to detect particular gene sequences in the amplified RNA virus and indiscriminately cleave nearby structures once complexed. While this technique is unique and excellent in terms of selectivity and sensitivity, it requires highly trained staff members and can only be utilized in laboratory-based hospitals where large stationary pieces of equipment, isotopes and reagents can be accessed easily.
In comparison, our NACT has several advantages over the alternative currently used techniques, as detailed above. Supplementary Table 4 shows a comparison of currently available COVID-19 detection techniques as compared with our developed protocol. Though several other isothermal NAA-based (especially RT-LAMP-based) colorimetric tests have been used for SARS-CoV-2 detection previously23,24, these relied on the use of pH-responsive dyes to indicate the successful amplification of the target sequence. While such dye-based NAA detection approaches offer sensitive and precise results, owing to the prevalence of spurious amplification products, the process also has a high occurrence of false-positive results64. Thus, the uniqueness of our protocol resides in the usage of ASO-capped AuNPs as a nanomaterial reporter that acts as a colorimetric indicator (Figs. 1 and 2) to address the limitation of false color changes induced by spurious NA amplification products when using pH-responsive dyes.

The collected sample is added to the lysis buffer and undergoes an isothermal NAA by incubation at 65 °C for 30 min. The amplification products are added to the Au-ASOTEST solution (containing the ASO-capped Au nanoparticles) for the clinical diagnosis of COVID-19 simply by observing the color change. Positive COVID-19 samples show color change from pink to purple, whereas negative samples remain pink.
The reliability of many diagnostic tests fails when they are tested either to detect a viral load at its early representation or to detect a viral gene mutated during its current spread65,66. Our sensor design attempts to compensate for these drawbacks by directly targeting the N gene of SARS-CoV-2 using specifically designed ASOs at multiple positions of the gene6,7 (Fig. 3), ensuring accurate recognition of the target sequence even if the other genomic segments of the virus are subject to mutation. Most of the known mutations for SARS-CoV-2 to date are single-point mutations. Mutations in any other region not targeted by the ASOs will not hamper the assay performance. Furthermore, even if there is a mutation in the RNA sequence targeted by the ASOs, a decrease in target binding energy for one of the ASOs may be observed, but due to our dual-targeted approach, the aggregation among the closely spaced gold nanoparticles should not be altered substantially. Detection of low viral loads at an early stage of infection will be directly related to the detection limit of the diagnostic assay, which is 10 copies/µl for this protocol.

Schematic representation of the hybridization process of the ASO-conjugated AuNPs to the complementary target gene sequence to enable colorimetric, naked-eye detection of SARS-CoV-2.
Experimental design
Sample preparation
Sample preparation for our NACT protocol is context dependent and needs to be adjusted to satisfy the specific environmental requirements. The choice of sampling technique will depend upon the test settings. Samples can be used directly, without prior RNA extraction, which is useful, for example, in low-resource settings or in rural and remote areas where basic laboratory equipment such as centrifuges may not be accessible, as well as in POC application of the protocol. Extraction and purification of RNA can, however, optionally be carried out under suitable laboratory settings, using commercially available kits. Column-magnetic bead and microbubble-based technologies are examples of RNA extraction methodologies67,68; however, specialized equipment, power and trained staff are required for RNA isolation and purification.
In our previous work, we extracted and purified RNA from Vero cells infected with either SARS-CoV-2 or Middle East respiratory syndrome (MERS)-CoV6,7. Also, RNA was extracted from confirmed positive and negative COVID-19 clinical samples (nasal and oropharyngeal swabs)7. Briefly, we used a column-based kit to isolate high-quality total RNA from samples in 20 min using a standard laboratory centrifuge. The kit utilizes guanidine-isothiocyanate lysis buffer to protect the RNA during the isolation steps. This also includes RNase-free lysis and wash solutions that protect RNA from RNases while liberating the RNA from DNA, proteins and other cellular debris. It also ensures the removal of the majority of genomic DNA. The process can typically be completed in less than 20 min without the need for hazardous phenol/chloroform extraction, CsCl centrifugation, or LiCl or alcohol precipitation. We have tested RNA extracted from positive COVID-19 samples, negative samples or Vero cells infected with MERS-CoV in addition to RNA spiked into artificial saliva. Ninety samples of spiked saliva samples were prepared and used for the protocol evaluation (see Supplementary Table 4 for further details).
RNA-extraction-free sample processing, however, requires no specialized equipment or trained staff. We therefore subsequently also investigated the performance of the NACT protocol with direct sampling, i.e., using an RNA-extraction-free approach. Guanidinium isothiocyanate lysis buffer at a sample: lysis buffer: water ratio of 2:1:2 was used for direct sample testing. We used this sample preparation condition because of its superior results compared with other tested sample preparation methods (Supplementary Table 5).
Design of NAA primers
We targeted the nucleocapsid phosphoprotein (N) gene sequence (1,260 bp) of SARS-CoV-2 isolates (2019-nCoV/USA-WA1-A12/2020); the accession number is MT020880 in the NCBI repository. The nucleotide sequence of the N gene ranges from 28,274 to 29,533, and the nucleotides selected for designing NAA primers were 28,321–29,520 (Supplementary Data 1). These 1,200 nucleotide bases were added to the PrimerExplorer v.5 (https://primerexplorer.jp/e/) software to design NAA primers suitable for the current NACT method22. In our previous publication, we identified four unique ASOs targeting particular segments of the N gene6,7. We observed that two of the developed ASOs were specific for nucleotides 28,695–28,736 (ASO1 and ASO2). The binding energy of this pair (i.e., ASO1 and ASO2) was found to be highest among other predicted pairs. Thus, NAA primers were designed to target the region covered by ASO1 and ASO2. Primers are typically designed with melting temperatures between 54 °C and 67 °C. Five NAA primer sets were generated that varied in their start and end positions within the targeted region. Among the generated five primer sets, we selected the set having dG = −2.36, which targets the 28,525–28,741 nucleotide range of the N-gene sequence. dG is the reaction’s disassociation constant, which measures the strength or spontaneity of dimerization.
RT-LAMP is a one-pot isothermal NAA method that first converts RNA to DNA, followed by amplification of cDNA in the same reaction pot. This NAA method therefore utilizes three pairs of primers and enzymes for NA amplification in a single tube. Briefly, the conversion of RNA to cDNA first takes place by the action of the reverse transcriptase enzyme. Four primers, that is, the forward inner primer (FIP), backward inner primer (BIP), forward outer primer (F3) and outer backward primer (B3), are then used to target four different regions of the gene, and Bst DNA polymerase is used to amplify NA under isothermal conditions, while the other two primers, known as ‘loop primers’ are used to accelerate the NAA reaction69,70. Thus, six primers are used under isothermal conditions to amplify a single target gene in this NAA technique, in contrast to RT-PCR, where primarily two primers are used at varying temperature conditions. Although this NAA produces many amplicons with various structural conformations instead of one major amplified genetic segment like RT-PCR, it has other advantages such as the use of cost-effective equipment and high levels of sensitivity and amplification efficiency compared with RT-PCR71. Herein, F3, B3, FIP and BIP sequences were identified to amplify the region targeted by ASO1 and ASO2. Forward loop (LF) and backward loop (LB) primers were also designed from these primers (Fig. 4). A primer set consisting of six primers has thus been designed for NAA, which are specific for the SARS-CoV-2 N-gene segment as presented in Table 1. It is important to note that primers can be designed to make the protocol applicable to any viral or bacterial sequence simply by altering the initial target nucleotide sequence. Currently, as a proof-of-concept, we have established the method to target the N-gene sequence of COVID-19 causative virus, SARS-CoV-2. The successful amplification of the SARS-CoV-2 RNA using this approach has been confirmed and optimized using gel electrophoresis (Fig. 5), which shows that the produced amplicons are of various structural conformations. The laddered banding pattern indicates positive amplification and spurious amplification products generated during the RT-LAMP reaction. This is a typical electrophoresis pattern seen with RT-LAMP and reported widely23,72. The NAA was carried out with primer concentrations as follows: FIP (16 µM), BIP (16 µM), F3 (2µM), B3 (2 µM), Loop F (4 µM) and Loop B (4 µM).

The SARS-CoV-2 N gene-specific NAA primer set (Table 1) has been designed to selectively amplify the 28,525–28,741 nucleotide segment. Their corresponding positions are demonstrated.

The six primers were added to the reaction mixture where the NA amplification takes place at 65 °C for 30 min. Lanes 2–8 show the amplification products using different concentrations of SARS-CoV-2 RNA, which appear as multiple bands of different molecular weights. DNA ladder and the negative control (water as a template) are shown in lane 1, 9 and 10, respectively. The gel results show the successful amplification of the genetic material.
Design and functionalization of ASOs
Software for statistical folding of NA and studies of regulatory RNAs, Soligo6,7,73, was used to design the ASOs targeted towards the N-gene sequence of SARS-CoV-2. The folding temperature was maintained at 37 °C, and ionic condition was kept at 1 M sodium chloride for a preferred ASO length of 20 nucleotide bases. The following parameters were used:
-
1
GC% should be within 40–60%.
-
2
Target sequences with GGGG will be eliminated.
-
3
Average unpaired probability of the ASOs for target site nucleotides is set to ≥0.5.
-
4
All sites targeted to the peak in the accessibility profile are ranked by their average unpaired probability (the higher the better). The threshold probability is >0.5.
-
5
Of those satisfying the above criteria 1–4, only the top 20 will be considered with the highest average unpaired probability.
-
6
Lastly, the binding energy of the ASOs was compared with the target sequence, and the binding energy cutoff for the selection of ASOs was kept at ≤−8 kcal/mol.
Among the 20 predicted ASO sequences, two of the ASOs were selected on the basis of their comparative binding disruption energies and binding energies with the target sequence ranging from nucleotides 28,695–28,736 of the N gene of SARS-CoV-2 (Table 2). The other important criterion for which these particular ASO sequences have been selected is their targeting ability to the genetic segment, which will be amplified by the designed primer sets. One of the other parameters behind the selection of these two ASO sequences was the proximity of their target positions6,7, as can be observed from Table 2. This facilitates agglomeration of gold nanoparticles selectively in the presence of the N-gene sequence of SARS-CoV-2 (Fig. 3). Towards this aim, the ASOs were differentially functionalized at either the 3′ or 5′ end. ASO1 and ASO2 were functionalized at the 5′ and 3′ end, respectively, with thiol moiety. Functionalizing one ASO at the 5′ end and another at the 3′ end upon their conjugation with AuNPs means that the nanoparticles will come close to each other when bound to their target RNA sequence. This will lead to the anticipated agglomeration among the nanoparticles and, hence, the change in plasmonic response (Supplementary Figs. 1 and 2).
Table 2 Selected ASO sequences targeted for the N gene of SARS-CoV-2 The efficiency of the kit depends on the design of the NAA primers (inner, outer and loop primers) and the ASOs. The screening and designing of the primers and ASOs are critical, because finding the right combination of ASOs that can specifically target only the amplified genetic segment is challenging. The best targeting ASO might lie in a region that is covered by nonspecific primers. Thus, a trade-off between the binding efficacy of the ASOs and the primers’ specificity should be made to select the optimal combination. We have demonstrated that targeting the amplified genetic segment with suitable ASOs underlies the kit’s accuracy, sensitivity and specificity6.
Preparing ASO-capped gold nanoparticles
These ASOs were used to cap gold nanoparticles6,7 using the capping agent exchange process. Gold nanoparticles were initially stabilized with sodium citrate and then exchanged (i.e., replacing the capping agent) with thiol-modified ASOs. Due to the higher binding energy of the Au-S bond, there is a preferential metal sulfur coordination bond between the thiol-modified ASOs to the AuNPs74. This replaces the citrate moieties with thiolated ASOs.
Readout of the NACT methodology
The NACT assay for SARS-CoV-2 detection utilizes AuNPs capped with ASO as a colorimetric reporter. The genetic material obtained from the samples is first amplified isothermally at 65 °C for 30 min. The ASO-capped AuNPs (an equivalent mixture of ASO1 capped AuNPs and ASO2 capped AuNPs) are then added to the amplified mixture. Due to the selectivity of the ASOs, the separately tagged AuNPs bind to the amplified genetic segments, leading to the agglomeration of AuNPs. This causes a change in the plasmonic signal such that the visible color of the solution changes from pink to purple only in the presence of the target genetic sequence. The visual readout of the test can be monitored by the naked eye (Figs. 6a, 7a and 8a). Further, the assay can be easily performed in a multiwell plate format, and the absorbance can be recorded using a conventional plate reader (Figs. 6b, 7b and 8b). The assay results can be also monitored using a handheld readout (e.g., portable plate reader). Figure 8 depicts a ‘clear color change’ that takes place in positive viral samples when samples are used directly, without RNA extraction. To avoid any ambiguity in interpreting the color of the sample, the protocol can also use a quantitative absorption measurement to avoid any subjectivity in the analysis.

a, The test mixture changes color from pink to purple as a response to SARS-CoV-2 RNA. Negative samples showed no obvious color change. b, Absorbance of the reaction mixture for 61 samples (29 positive and 32 negative) was recorded using a plate reader. COVID-19 samples were initially tested using FDA-approved RT-PCR kit (LABGUN), and the findings were plotted against our NACT results, which corroborated the RT-PCR findings. Threshold line has been set that maximizes the assay performance.

a, The test mixture changes color from pink to purple as a response to SARS-CoV-2 RNA. Negative samples showed no obvious color change. No cross-reactivity was observed in the test results from the samples containing MERS-CoV RNA. b, Absorbance of the reaction mixture for 60 positive and 30 negative samples was recorded using a plate reader. The sample number represents the numbering of the tested samples. A threshold line has been set that maximizes the assay performance.

a, Photographic images of tubes with 22 samples (11 positives and 11 negatives) treated with guanidium isothiocyanate (GITC) lysis buffer and tested according to our RNA-extraction-free NACT protocol. SARS-CoV-2 N-gene targeted ASO-conjugated Au nanoparticles showed a clear color change for the positive viral samples but not for the negative samples. b, Absorbance of the reaction mixture for 22 samples (11 positives and 11 negatives) were recorded using a plate reader. COVID-19 samples were initially tested using FDA-approved RT-PCR kit (LABGUN), and the findings were plotted against our NACT results, which corroborated the RT-PCR findings. Threshold line has been set that maximizes the assay performance.
Qualitative and semiquantitative detection
The first line of COVID-19 management is to separate and quarantine the infected people from their surroundings as soon as possible. Therefore, it is sufficient to distinguish between the presence and absence of SARS-CoV-2 RNA and thus identify the infected samples. Thus, the qualitative detection of COVID-19 is far more important than the quantitative detection of the viral load in the infected samples. However, the aggregation of ASO-capped AuNPs leading to their change in surface plasmon bands and change in UV-visible absorbance can be quantitative. However, the color change of the solution can only be used to provide a Yes/No answer. Like most naked-eye colorimetric approaches, NACT also depends on the user’s observation and color perception to draw inference on the diagnosis of the sample (i.e., positive or negative). Therefore, the introduction of a plasmonic optical readout will further eliminate the subjectivity of the test and avoid potential errors due to the variation in color interpretation from person to person. Additionally, the test could be made quantitative by establishing a standard curve that correlates the viral RNA quantity with an optical readout.
Although herein we have represented our data only in terms of increase in absorbance at 660 nm wavelength (Figs. 6–8), there is a substantial red shift in the absorbance spectra of the gold nanoparticles in the presence of SARS-CoV-2 RNA. A shift of ~40 nm was observed in the aggregation band of ASO-conjugated AuNPs only in the presence of the SARS-CoV-2 RNA6,7, as shown in Supplementary Fig. 1. Hence, there will be no problem in discriminating between the positive and negative samples by the naked eye. The aggregation of ASO-capped AuNPs was investigated not only through UV-visible absorbance spectroscopy but also through Zetaview experiments, transmission electron microscopy and hyperspectral imaging (HSI) to support the agglomeration phenomena6,7. Where UV-visible absorbance demonstrated the shift in the surface plasmon band of the AuNPs upon selective detection of the genetic material, Zetaview showed an increase in the size of the nanoparticles upon their attachment with the target site6. Upon the addition of the target RNA, various clustering entities were observed under transmission electron microscopy6. The presence of the target RNA was found to form clusters of ~80–120 nm entities (Supplementary Fig. 2), indicating the interparticle aggregation mediated by intermolecular hydrogen bonding among the nucleotides6,7. The formation of the large entities was further explored using HSI to localize the ASO-tagged nanoparticles. HSI studies further support the formation of large particle clusters due to the binding of the ASOs to their target region6,7. All this points towards a multiparticle aggregation phenomenon caused by intermolecular hydrogen bonding among the nucleotides, which also influences the agglomeration among multiple gold nanoparticles to form a clustered assembly.
Avoiding contamination
To avoid any unexpected outcome of the test results, the ASOs and ASO-capped AuNPs were suspended in RNase and DNase-free water in each of the preparation steps. Proper sterile conditions were maintained especially during the NAA step. To do so, the biosafety cabinet and the surrounding environment were cleaned properly with 70% (vol/vol) ethanol and RNaseZap solution each time before and after use. Pipettes and other bottles were also cleaned each time to optimize the test condition by removing any unexpected contamination.
Validations and controls
The negative control must be water only, whereas synthetic RNA containing the target segment can be used as a positive control. The validity of the NACT approach can be evaluated by testing the specificity of AuNPs capped with ASOs using a sample with the SARS-CoV-2 gene sequence but lacking the target N-gene sequence targeted by the ASOs. In the absence of the target genetic strand, the color of the solution did not change. This indicates a loss in sensing efficiency of the ASO-capped AuNPs in the absence of the desired SARS-CoV-2 N-gene strand, inhibiting the change in color of the solution (Fig. 9).

a, Color of the NACT test remains pink as a response to the negative control (water) and synthetic SARS-CoV-2 RNA lacking the targeted N-gene segment; change in color to purple observed in response to SARS-CoV-2 RNA containing the target sequence. b, Zoomed image of a.
Adaptability of the NACT protocol to test for other pathogens
To adapt the protocol to other targets, the following steps would be necessary:
-
1
Design new ASOs. Suitable ASO sequences need to be designed that specifically target RNA of the disease-causing virus/pathogen. Particular attention must be paid to designing the ASO sequences since their proximity will influence the agglomeration of the nanoparticles leading to a color change (Fig. 3).
-
2
Design new primers. Next, the inner, outer and loop primer sequences need to be altered to amplify the target gene sequence to which the ASOs will hybridize. Overall, by changing only the sequences of ASOs and primers, the capability of the protocol could be expanded to other pathogens.
-
3
Standardization of lysis buffer. Suitable standardization of lysis buffers must be performed while utilizing this technology for the detection of other targets. The membranes of different microorganisms comprise varied biological materials and, hence, might behave differently in different types of lysis buffer. Therefore, to obtain an optimized result, one needs to standardize the lysis conditions for each target as was done during the development of this protocol for SARS-CoV-2.
Expertise needed to implement the protocol
Basic knowledge in molecular biology is required to design ASOs and NAA primers. Basic knowledge of material science and chemistry is also recommended to perform Steps 1–7, the synthesis of AuNPs and their conjugation with ASOs. Once all the reagents and required materials are prepared, performing the test is quite straightforward. As our protocol offers the possibility of COVID-19 testing even without the extraction of RNA, it enables individuals with limited scientific expertise to conduct the test.
Materials
Biological materials
-
Deidentified clinical samples having positive or negative SARS-CoV-2 infection (nasal swab, nasopharyngeal swab and oropharyngeal swab)
Caution
Biosafety regulations should be followed while handling any potentially infectious clinical samples. In the case of any uncertainty, contact your local safety officer. We obtained permission from the Institutional Biosafety Committee (IBC) for handling these infectious samples as mentioned herein through the IBC submission number IBC-00005683
Caution
Users handling infectious biological materials should undergo requisite training and take necessary care to minimize the potential risk. Necessary personal protective equipment (PPE) should always be worn while working in the laboratory with these samples. Necessary care must be taken to destroy the hazardous materials before they come into contact with the environment. The materials should either be treated with proper disinfectants, acids or bases. The work surfaces should also be decontaminated with 70% (vol/vol) ethanol and disinfectants, wherever necessary. In the event of an exposure, the contaminated surfaces should be cleaned with soap and water, and exposed mucus membranes should be flushed with water. Environmental Health and Safety must be contacted, and the first report of injury should be completed. Personnel should also seek medical care at the appropriate center.
Reagents and consumables
-
Water, molecular biology grade, nuclease protease-free and low endotoxin, sterile, 1 L (Quality Biological, cat. no. 351-029-131)
-
ASO strands (GenScript, custom-made sequences where either of the 5′- or 3′-ends are functionalized with thiol moieties; see Table 2)
Critical
The current study used thiolated ASOs for the conjugation with citrate-stabilized gold nanoparticles. However, it should be noted that the oligos are easily oxidizable during shipping and storage to form disulfide linkages. Hence, the disulfide bonds need to be broken before functionalization with gold nanoparticles either by treatment with DTT or Tris(2-carboxyethyl)phosphine, as detailed by the oligo manufacturer.
-
Primers for NA amplification step (GenScript, custom made sequences for inner, outer and loop primers; see Table 1)
-
Quantifluor ssDNA System (Promega, cat. no. E3190)
Caution
This reagent needs to be stored at −20 °C for its stability. The dye should be protected from long light exposure.
-
Purelink RNA mini kit (Invitrogen, cat. no. 12183018A); The lysis buffer provided with this kit is used as the lysis buffer containing guanidinium isothiocyanate for direct sampling (Step 9B).
Caution
The kit can be stored at room temperature (25 °C). Be careful about possible contamination during handling.
-
Quick-RNA viral kit (Zymogen, cat. no. R1034)
Caution
The kit can be stored at room temperature. Be careful about possible contamination during handling.
-
Warmstart Lamp Kit (DNA and RNA), 500 Rxns (New England Biolabs, cat. no. E1700L)
Critical
The current protocol used the WarmStart LAMP Kit, procured from New England Biolabs, to benefit from the WarmStart technology and inhibit the activity of the NAA reagents at room temperature
Caution
The kit should be stored at −20 °C in separate aliquots to avoid the repeated freeze–thaw cycle of the stock.
-
Tetrachloroauric(III) acid trihydrate (Sigma, cat. no. 1015820001)
Caution
The reagent should be protected from long exposure to moisture. A stock solution having a high concentration of HAuCl4 can be prepared in water and used as and when necessary.
-
Sodium citrate tribasic dihydrate (Sigma, cat. no. S4641)
Caution
During the preparation of gold nanoparticles, sodium citrate solution should always be prepared fresh. The use of old stock will hamper the preparation of AuNPs.
Equipment
-
Microcentrifuge tubes, 0.2 ml (Sigma, cat. no. Z374873), 0.5 ml, 1.5 ml and 2 ml capacity (USA Scientific, cat. no. 1605-0000, 1615-5510 and 1620-2700, respectively)
-
Micropipette tips, 10 µl, 200 µl and 1 ml tips (USA Scientific, cat. nos. 1180-3710, 1120-8710 and 1182-1730, respectively)
-
Microamp Fast Optical 96-well Reaction Plate, 0.1 ml (Invitrogen, cat. no. 4346907)
-
Centrifuge conical tube, 50 ml (Fisher Scientific, cat. no. 1495949A)
-
Centrifuge conical tube, 15 ml (Fisher Scientific, cat. no. 1495970C)
-
Low background glass scintillation vials, 20 ml, poly cap (Research Product International, cat. no. 121000CA)
-
Benchtop Centrifuge (Eppendorf, cat. no. 5804)
-
Thermomixer, 24-tube capacity (Eppendorf, model no. 5355 30794)
-
Magnetic hotplate stirrer (IKA, model no. C-Mag HS4)
-
Magnetic spin bar micro-stir bar, 15 × 79 mm (VWR, cat. no. 58948-353)
-
Fisherbrand Elite adjustable-volume pipettes, 0.5–5 µl, 2–20 µl, 20–200 µl and 100–1,000 µl (Fisher Scientific, cat. nos. FBE00005, FBE00020, FBE00200 and FBE01000 respectively)
-
Bath sonicator (Branson 2800, model no. CPX 2800)
-
Weighing balance (VWR, model no. VWR-124B2)
-
GelDoc imager (BioRad, model no. GelDoc EZ)
-
Particle tracking analyzer (Zetaview, model no. PMX-220-12C-R3)
-
pH-meter (HACH, model no. HQ440D multi)
-
Biotek Synergy Neo2 microplate reader (Biotek, model no. 1909175)
-
Yamato PiCOEXPLORER photo absorbance meter (Yamota, model no. PAS-110-YU)
-
Freezer, 4 °C (Frigidaire, model no. R134A; ASF35BU6)
-
Freezer, −20 °C (Thermo Fisher Scientific, model no. 20LFEEFSA)
-
Freezer, −80 °C (Haier, model no. DW-86L728JA)
Software
-
Gel quantification software (Bio-Rad ImageLab)
-
PrimerExplorerV5 (https://primerexplorer.jp/e/)
-
Sensitivity, specificity and accuracy has been calculated using MedCalc Free statistical calculators (https://www.medcalc.org/calc/diagnostic_test.php) based on the following equations:75,76,77,78,79,80
-
Accuracy is the total number of samples correctly diagnosed over the total number of tested samples. The accuracy can be calculated using the following equation:
$$=\frac{{{\mathrm{True}}\,{\mathrm{positive}} + {\mathrm{True}}\,{\mathrm{negative}}}}{{{\mathrm{True}}\,{\mathrm{positive}} + {\mathrm{True}}\,{\mathrm{negative}} + {\mathrm{False}}\,{\mathrm{positive}} + {\mathrm{False}}\,{\mathrm{negative}}}}$$ -
Sensitivity represents the probability of having a positive test result given the disease is present, true positive rate (TP)
$$= \frac{{{\mathrm{True}}\,{\mathrm{positive}}}}{{{\mathrm{True}}\,{\mathrm{positive}} + {\mathrm{False}}\,{\mathrm{negative}}}}$$ -
Specificity represents the probability of having a negative test result given the subject is healthy, true negative rate (TN)
$$= \frac{{{\mathrm{True}}\,{\mathrm{negative}}}}{{{\mathrm{True}}\,{\mathrm{negative}} + {\mathrm{False}}\,{\mathrm{positive}}}}$$
Reagent setup
-
Tetrachloroauric(III) acid trihydrate, 1 mM: dissolve 1 g of tetrachloroauric(III) acid trihydrate in 250 ml of distilled water to make a 10 mM stock solution. Dilute the 10 mM stock with RNAse-free water to prepare a 1 mM solution
-
Sodium citrate solution, 1% (wt/vol): dissolve 0.5 g of sodium citrate in 50 ml of distilled water to prepare 1% sodium citrate solution
Critical
The sodium citrate solution should be prepared fresh each time.
Procedure
Synthesis of gold nanoparticles
Timing 1 h
-
1
Heat 20 ml of 1 mM HAuCl4·3H2O solution to boil under stirring conditions, and then add 2 ml of 1% citrate solution.
Caution
The gold nanoparticle solution should not be stirred for more than 10 min.
-
2
Remove the mixture from heat after 10 min.
Critical step
The color of the solution should change to wine red within this time.
-
3
Centrifuge 10 ml of the gold nanoparticle solution at 25,000g for 30 min at room temperature 10 ml and discard the supernatant.
-
4
Redisperse the pellet in 1 ml of RNase-free water with mild vortexing.
Pause point
If needed, the citrate-stabilized gold nanoparticles can be stored at room temperature for future use, but the solution should be used within 7–10 d of its preparation.
Capping of AuNPs with ASOs
Timing 1 h
-
5
Functionalize two 1 ml aliquots of gold nanoparticles suspended in RNase-free water (from Step 4) by adding 5 µl of ASO1 or 2.5 µl of ASO2 to each, respectively, in separate centrifuge tubes under stirring at 1,500g for 30 min at room temperature.
-
6
Prepare a unique cocktail of ASO-conjugated Au nanoparticles (Au-ASOmix solution) by mixing an equivalent ratio of the two Au-ASO nanoparticle solutions. For example, to prepare 1 ml of Au-ASOmix solution, add 500 µl of Au-ASO1 solution to 500 µl of Au-ASO2 solution.
-
7
To prepare the Au-ASOmix test solution (Au-ASOTEST), dilute the Au-ASOmix solution by adding 80 µl of this Au-ASOmix solution to 920 µl of RNase-free water.
Pause point
Au-ASOTEST can be stored at 4 °C for future use and can be used for more than 3 months.
Collection of samples
Timing 2 min
-
8
Collect clinical samples (i.e., nasal and oropharyngeal swabs and/or saliva) from the patients under observation with suspected COVID-19.
Critical step
Handle samples with necessary PPE.
Pause point
The samples can be stored for at least 6 months at −80 °C.
Sample preparation
Timing 30 min
-
9
The collected clinical sample can be prepared by either extracting the RNA from the sample (using option A) or treating samples directly without RNA extraction by lysing the sample directly (option B). The selection of the sample preparation method is user-dependent based on the test settings (see Experimental design for further information).
-
(A)
RNA extraction from the samples
-
(i)
Isolate the total RNA from the samples using the Invitrogen PureLink RNA mini kit by following the manufacturer’s instrucutions81.
Pause point
The sample can be stored in different aliquots at −80 °C in RNase-free centrifuge tubes.
-
(i)
-
(B)
Direct sample treatment with lysis buffer
-
(i)
Add 4 µl of the clinical samples (as they are, without any prior processing) to 2 µl guanidinium isothiocyanate lysis buffer (from Purelink RNA mini kit) with 4 µl of RNase-free water and mix by tapping the tube gently.
Pause point
The direct sample should be prepared fresh each time.
-
(i)
-
(A)
NAA step of the targeted segments
Timing 35 min
Critical
The NAA step is an extremely sensitive amplification method23,25. Care should be taken when testing multiple samples simultaneously to avoid contamination. Reaction containers should be opened and handled in a secondary location away from the RNA-extraction location and equipment. Regular decontamination of the working place is also important to reduce the risk of contamination. We also suggest performing positive and negative controls alongside the experiment each time (see Experimental design).
-
10
Prepare the preamplification space by wiping the surface with 70% (vol/vol) ethanol and RNase Zap solution.
-
11
Prepare the following mixture of NAA primers mix (10×):
Component
Final concentration (µM)
Amount (µl)
FIP (100 µM)
16
32
BIP (100 µM)
16
32
F3 (100 µM)
2
4
B3 (100 µM)
2
4
Loop F (100 µM)
4
8
Loop B (100 µM)
4
8
RNase-free water
112
-
12
Prepare the following NAA reaction mixture in a 1.5 ml Eppendorf or PCR tube (enough for four replicates):
Component
Amount (µl)
WarmStart LAMP 2X Master Mix
5
LAMP Primer Mix (10×)
1
RNase-free water
2
Total volume
8
Critical step
Use WarmStart technology to inhibit activity of the NAA reagents at room temperature. Briefly, the kit includes WarmStart LAMP 2X Master Mix, which contains a blend of Bst 2.0 WarmStart DNA Polymerase and WarmStart RTx Reverse Transcriptase in an optimized LAMP buffer solution.
-
13
Prepare 8 μL aliquots of the NAA reaction mixture from Step 12, and add to 2 μL of sample (from Step 9) in PCR strip tubes or PCR plates. Mix well by pipetting up and down a few times (sample can be either extracted RNA or direct sample treated with lysis buffer or artificial saliva sample spiked with clinical samples’ RNA). Add water separately to the NAA reaction as a negative control.
-
14
Perform the reactions at 65 °C for 30 min on a heat block.
-
15
Inactivate the Bst 2.0 polymerase and reverse transcriptase enzyme (RTx) by heating the reaction mixture at 85 °C for 5 min. Confirm the successful amplification of the target using gel electrophoresis82 as shown in Fig. 5.
Critical step
It is recommended to perform the NA detection (Step 16) immediately after the NAA step.
Pause point
Amplified cDNA can be kept on ice and used within 1 h.
Colorimetric detection
Timing 5 min
-
16
Add 100 µl of Au-ASOTEST (from Step 7), to the tube containing the 10 µl of the amplified gene. Heat the mixture at 65 °C for 5 min.
-
17
The color change can be observed with the naked eye, and/or by a plate reader. In the latter case, transfer the mixture to a 96-well-plate and spectroscopically investigate the color change by reading the absorbance spectrum at 660 nm.
Troubleshooting
The problems observed during the testing of this protocol are listed in Table 3.
Timing
-
Steps 1–7, preparation of ASO-capped gold nanoparticles (Au-ASO): 2 h
-
Steps 8–9, collection of samples for direct use or after RNA extraction and purification: ~30 min
-
Steps 10–15, isothermal amplification of the target genetic material: 35 min
-
Steps 16–17, colorimetric detection of the target genetic material: 5 min
Anticipated results
In ref. 7 we demonstrated the successful use of our NACT technology to rapidly and selectively diagnose COVID-19 using 48 nasopharyngeal swab clinical samples with nearly 100% accuracy, sensitivity and specificity. This modified protocol has shown 98.4% accuracy, 96.6% sensitivity and 100% specificity using 61 nasopharyngeal swab clinical samples (M.A., P.M., K.D. and D.P., unpublished results, Fig. 6). Based on our findings, the NACT works on a variety of biological samples, including nasopharyngeal, oropharyngeal swabs7 and artificial saliva (M.A., P.M., K.D. and D.P., unpublished results). Future studies are required to test the performance of the test kit on saliva samples collected from COVID-19 infected patients83,84. The protocol was successful in distinguishing artificial saliva samples spiked with positive COVID-19 samples’ RNA from the negative ones. Samples spiked with RNA from COVID-19 samples confirmed positive using RT-PCR COVID-19 LABGUN kit (EUA FDA approved) showed a prominent color change from pink to purple, except one (Fig. 7). This sample from the positive set did not change color owing to the inappropriate sealing of the tube cap. This caused evaporation in the liquid mixture during the NAA step at 65 °C, leading to the eventual false-negative results. Hence this false-negative test point can be assigned to human handling error. Samples spiked with MERS-CoV RNA or RNA from COVID-19 negative samples showed no color change (pink).
Our protocol enables minimal sample processing and works with both direct samples (RNA-extraction-free approach) and RNA extracted from clinical samples. The RNA-extraction-free protocol takes <40 min to complete, and the results can be interpreted qualitatively (Yes/No) as a visual color change from pink (negative) to purple (positive), as demonstrated in Fig. 1. A time estimate of the whole process and each individual step is also shown in Supplementary Table 3. In total, 22 direct samples (11 SARS-CoV-2 positives and 11 negatives) have been tested using our NACT assay, and the results are highlighted in Fig. 8. Our NACT method can accurately determine SARS-CoV-2 positive and negative clinical samples even without the extraction of RNA. The confusion matrix benchmarking the results against the gold standard RT-qPCR can be seen in Supplementary Fig. 3.
To avoid errors due to the variation in color interpretation among individuals, the NACT can also be coupled with a portable handheld reader or a standard plate reader for quantitative analysis (Figs. 6–8). The optical density-based readout system can reduce human error by recording the absorbance of the sample and identifying them as positive or negative using a set threshold value, which has been selected carefully using the receiver operating curve to optimize the sensor performance, as shown in Supplementary Fig. 4. Using threshold values of 0.19 for the RNA-extracted samples7 and 0.152 for the direct samples, the test was found to have sensitivity of 96.6%, or 100%, respectively, and specificity of 100%, or 100%, respectively (M.A., P.M., K.D. and D.P., unpublished results, see Supplementary Table 1), with a detection limit of 10 copies/μL. It is worth noting that the estimation of the sensitivity and specificity of an assay often needs evaluation with a large pool of samples. The values determined for the NACT test are likely to be dependent on the sample size, and hence, the >99% specificity is only claimed for the given number of nasal swab samples tested in this study. The protocol successfully diagnosed COVID-19 from several clinical samples with accuracy, sensitivity and specificity as high as those of the results from gold-standard RT-qPCR.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting all the findings of this study are available within the article and Supporting Information files. The primary research articles5,6,7 support the protocol development and use for colorimetric detection of SARS-CoV-2. This includes the ASO design and optimization, the AuNPs capping and detection of SARS-CoV-2 using genomic RNA samples, RNA extracted from Vero cells infected with SARS-CoV-2, and nasopharyngeal swab samples. The cross-reactivity studies and benchmarking data using RT-qPCR are also included in our previous reports for the protocol carried out using RNA extraction7. Source data are provided with this paper. All the other relevant data are available from the authors upon reasonable request
References
Minchin, S. & Lodge, J. Understanding biochemistry: structure and function of nucleic acids. Essays Biochem. 63, 433–456 (2019).
Godri Pollitt, K. J. et al. COVID-19 vulnerability: the potential impact of genetic susceptibility and airborne transmission. Hum. Genomics 14, 17 (2020).
Mason, M. G. & Botella, J. R. Rapid (30-second), equipment-free purification of nucleic acids using easy-to-make dipsticks. Nat. Protoc. 15, 3663–3677 (2020).
Suea-Ngam, A. et al. Enzyme-assisted nucleic acid detection for infectious disease diagnostics: moving toward the point-of-care. ACS Sensors 5, 2701–2723 (2020).
Moitra, P. et al. Rapid and low-cost sampling for detection of airborne SARS-CoV-2 in dehumidifier condensate. Preprint at medRxiv https://www.medrxiv.org/content/10.1101/2020.10.08.20208785v1 (2020).
Moitra, P., Alafeef, M., Dighe, K., Frieman, M. B. & Pan, D. Selective naked-eye detection of SARS-CoV-2 mediated by N gene targeted antisense oligonucleotide capped plasmonic nanoparticles. ACS Nano 14, 7617–7627 (2020).
Alafeef, M., Dighe, K., Moitra, P. & Pan, D. Rapid, ultrasensitive, and quantitative detection of SARS-CoV-2 using antisense oligonucleotides directed electrochemical biosensor chip. ACS Nano 14, 17028–17045 (2020).
Cantera, J. L. et al. Assessment of eight nucleic acid amplification technologies for potential use to detect infectious agents in low-resource settings. PLoS ONE 14, e0215756 (2019).
Yuan, X. et al. Current and perspective diagnostic techniques for COVID-19. ACS Infect. Dis. 6, 1998–2016 (2020).
Kabir, M., Afzal, M. S., Khan, A. & Ahmed, H. COVID-19 pandemic and economic cost; impact on forcibly displaced people. Travel Med. Infect. Dis. 35, 101661–101661 (2020).
Gautret, P. et al. International mass gatherings and travel-associated illness: a GeoSentinel cross-sectional, observational study. Travel Med. Infect. Dis. 32, 101504–101504 (2019).
Mishra, T. et al. Pre-symptomatic detection of COVID-19 from smartwatch data. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-020-00640-6 (2020).
Ding, X. et al. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11, 4711 (2020).
Vandenberg, O., Martiny, D., Rochas, O., van Belkum, A. & Kozlakidis, Z. Considerations for diagnostic COVID-19 tests. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-020-00461-z (2020).
Miesler, T., Wimschneider, C., Brem, A. & Meinel, L. frugal innovation for point-of-care diagnostics controlling outbreaks and epidemics. ACS Biomater. Sci. Eng 6, 2709–2725 (2020).
Talebian, S., Wallace, G. G., Schroeder, A., Stellacci, F. & Conde, J. Nanotechnology-based disinfectants and sensors for SARS-CoV-2. Nat. Nanotechnol. 15, 618–621 (2020).
Kosack, C. S., Page, A. L. & Klatser, P. R. A guide to aid the selection of diagnostic tests. Bull. World Health Organ. 95, 639–645 (2017).
Thompson, D. & Lei, Y. Mini review: recent progress in RT-LAMP enabled COVID-19 detection. Sens. Actuators Rep. 2, 100017 (2020).
Rabe, B. A. & Cepko, C. SARS-CoV-2 detection using isothermal amplification and a rapid, inexpensive protocol for sample inactivation and purification. Proc. Natl Acad. Sci. 117, 24450–24458 (2020).
Ganguli, A. et al. Rapid isothermal amplification and portable detection system for SARS-CoV-2. Proc.Natl Acad. Sci. 117, 22727–22735 (2020).
Farooq, U. et al. Loop-mediated isothermal amplification (RT-LAMP): a new approach for the detection of foot-and-mouth disease virus and its sero-types in Pakistan. Iran. J. Vet. Res. 16, 331–334 (2015).
Ahn, S. J. et al. Rapid and simple colorimetric detection of multiple influenza viruses infecting humans using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform. BMC Infect. Dis. 19, 676 (2019).
Dao Thi, V. L. et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 12, eabc7075 (2020).
Chow, F. W.-N. et al. A rapid, simple, inexpensive, and mobile colorimetric assay COVID-19-LAMP for mass on-site screening of COVID-19. Int. J. Mol. Sci. 21, 5380 (2020).
Xue, G. et al. Reverse-transcription recombinase-aided amplification assay for rapid detection of the 2019 novel coronavirus (SARS-CoV-2). Anal. Chem. 92, 9699–9705 (2020).
Khan, M. S. et al. Electrically-receptive and thermally-responsive paper-based sensor chip for rapid detection of bacterial cells. Biosens. Bioelectron. 110, 132–140 (2018).
Khan, M. S. et al. Detection of prostate specific antigen (PSA) in human saliva using an ultra-sensitive nanocomposite of graphene nanoplatelets with diblock-co-polymers and Au electrodes. Analyst 143, 1094–1103 (2018).
Khan, M. S. et al. Paper-based analytical biosensor chip designed from graphene-nanoplatelet-amphiphilic-diblock-co-polymer composite for cortisol detection in human saliva. Anal. Chem. 89, 2107–2115 (2017).
Srivastava, I. et al. On-chip electrical monitoring of real-time “soft” and “hard” protein corona formation on carbon nanoparticles. Small Methods 4, 2000099 (2020).
Li, H. & Rothberg, L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc. Natl Acad. Sci. USA 101, 14036–14039 (2004).
Liu, J. & Lu, Y. Accelerated color change of gold nanoparticles assembled by DNAzymes for simple and fast colorimetric Pb2+ detection. J. Am. Chem. Soc. 126, 12298–12305 (2004).
Wachiralurpan, S. et al. Rapid colorimetric assay for detection of listeria monocytogenes in food samples using LAMP formation of DNA concatemers and gold nanoparticle-DNA probe complex. Front. Chem. 6, 90–90 (2018).
Ventura, B. D. et al. Colorimetric test for fast detection of SARS-CoV-2 in nasal and throat swabs. ACS Sens. 5, 3043–3048 (2020).
Alafeef, M., Dighe, K. & Pan, D. Label-free pathogen detection based on yttrium-doped carbon nanoparticles up to single-cell resolution. ACS Appl. Mater. Interfaces 11, 42943–42955 (2019).
Alafeef, M., Moitra, P. & Pan, D. Nano-enabled sensing approaches for pathogenic bacterial detection. Biosens. Bioelectron. 165, 112276 (2020).
Ostadhossein, F. & Pan, D. Functional carbon nanodots for multiscale imaging and therapy. WIREs Nanomed. Nanobiotechnol. 9, e1436 (2017).
Misra, S. K. et al. Macromolecularly “caged” carbon nanoparticles for intracellular trafficking via switchable photoluminescence. J. Am. Chem. Soc. 139, 1746–1749 (2017).
Misra, S. K., Ostadhossein, F., Daza, E., Johnson, E. V. & Pan, D. Hyperspectral imaging offers visual and quantitative evidence of drug release from zwitterionic-phospholipid-nanocarbon when concurrently tracked in 3D intracellular space. Adv. Funct. Mater. 26, 8031–8041 (2016).
Mukherjee, P. et al. Tunable luminescent carbon nanospheres with well-defined nanoscale chemistry for synchronized imaging and therapy. Small 11, 4691–4703 (2015).
Datta, S. et al. Orthogonal self-assembly of an organoplatinum(II) metallacycle and cucurbit[8]uril that delivers curcumin to cancer cells. Proc. Natl Acad. Sci. 115, 8087–8092 (2018).
Srivastava, I. et al. Surface chemistry of carbon nanoparticles functionally select their uptake in various stages of cancer cells. Nano Res. 10, 3269–3284 (2017).
Misra, S. K. et al. 3D-printed multidrug-eluting stent from graphene-nanoplatelet-doped biodegradable polymer composite. Adv. Healthc. Mater. 6, 1700008 (2017).
Misra, S. K. et al. Trimodal therapy: combining hyperthermia with repurposed bexarotene and ultrasound for treating liver cancer. ACS Nano 9, 10695–10718 (2015).
Misra, S. K. et al. Regulating biocompatibility of carbon spheres via defined nanoscale chemistry and a careful selection of surface functionalities. Sci. Rep. 5, 14986 (2015).
Soodgupta, D. et al. Small molecule MYC inhibitor conjugated to integrin-targeted nanoparticles extends survival in a mouse model of disseminated multiple myeloma. Mol. Cancer Ther. 14, 1286–1294 (2015).
Misra, S. K., Jensen, T. W. & Pan, D. Enriched inhibition of cancer and stem-like cancer cells via STAT-3 modulating niclocelles. Nanoscale 7, 7127–7132 (2015).
Quemener, A. M. et al. The powerful world of antisense oligonucleotides: from bench to bedside. WIREs RNA 11, e1594 (2020).
Chan, J. H., Lim, S. & Wong, W. F. Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 33, 533–540 (2006).
Kher, G., Trehan, S. & Misra, A. Antisense oligonucleotides and RNA interference. in Challenges in Delivery of Therapeutic Genomics and Proteomics (ed. Misra, A.) 325–386 (Elsevier, 2011).
Smyrlaki, I. et al. Massive and rapid COVID-19 testing is feasible by extraction-free SARS-CoV-2 RT-PCR. Nat. Commun. 11, 4812 (2020).
Udugama, B. et al. Diagnosing COVID-19: the disease and tools for detection. ACS Nano 14, 3822–3835 (2020).
Carter, L. J. et al. Assay techniques and test development for COVID-19 diagnosis. ACS Cent. Sci. 6, 591–605 (2020).
Moitra, P., Bhagat, D., Pratap, R. & Bhattacharya, S. A novel bio-engineering approach to generate an eminent surface-functionalized template for selective detection of female sex pheromone of Helicoverpa armigera. Sci. Rep. 6, 37355 (2016).
Jayamohan, H. et al. SARS-CoV-2 pandemic: a review of molecular diagnostic tools including sample collection and commercial response with associated advantages and limitations. Anal. Bioanal. Chem. https://doi.org/10.1007/s00216-020-02958-1 (2020).
Lenartowicz, E. et al. Antisense oligonucleotides targeting influenza A segment 8 genomic RNA inhibit viral replication. Nucleic Acid Ther. 26, 277–285 (2016).
Shevtsov, M., Zhao, L., Protzer, U. & van de Klundert, M. A. A. Applicability of metal nanoparticles in the detection and monitoring of hepatitis b virus infection. Viruses 9, 193 (2017).
Parida, M., Sannarangaiah, S., Dash, P. K., Rao, P. V. L. & Morita, K. Loop mediated isothermal amplification (LAMP): a new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol. 18, 407–421 (2008).
Yu, L. et al. Rapid colorimetric detection of COVID-19 coronavirus using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform: iLACO. Preprint at medRxiv https://doi.org/10.1101/2020.02.20.20025874 (2020).
Yu, L. et al. Rapid detection of COVID-19 coronavirus using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform. Clin. Chem. 66, 975–977 (2020).
Qiu, G. et al. Dual-functional plasmonic photothermal biosensors for highly accurate severe acute respiratory syndrome coronavirus 2 detection. ACS Nano 14, 5268–5277 (2020).
Vogels, C. B. F. et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 5, 1299–1305 (2020).
El-Tholoth, M., Bau, H. H. & Song, J. A single and two-stage, closed-tube, molecular test for the 2019 novel coronavirus (COVID-19) at home, clinic, and points of entry. Preprint at chemRxiv https://doi.org/10.26434/chemrxiv.11860137 (2020).
Broughton, J. P. et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020).
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O. & Zhang, F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019).
Afzal, A. Molecular diagnostic technologies for COVID-19: limitations and challenges. J. Adv. Res. 26, 149–159 (2020).
Syal, K. Newly identified limitations of diagnostic tools for COVID-19 and consequences. J. Med. Virol. https://doi.org/10.1002/jmv.26673 (2020).
Kim Tiam, S. et al. Development of a new extraction method based on high-intensity ultra-sonication to study RNA regulation of the filamentous cyanobacteria Planktothrix. PLoS ONE 14, e0222029 (2019).
Klein, S. et al. SARS-CoV-2 RNA extraction using magnetic beads for rapid large-scale testing by RT-qPCR and RT-LAMP. Viruses 12, 863 (2020).
Mori, Y. & Notomi, T. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J. Infect. Chemother. 15, 62–69 (2009).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63–e63 (2000).
Zhang, G., Brown, E. W. & González-Escalona, N. Comparison of real-time PCR, reverse transcriptase real-time PCR, loop-mediated isothermal amplification, and the FDA conventional microbiological method for the detection of Salmonella spp. in produce. Appl. Environ. Microb. 77, 6495–6501 (2011).
Lamb, L. E., Bartolone, S. N., Ward, E. & Chancellor, M. B. rapid detection of novel coronavirus (COVID-19) by reverse transcription-loop-mediated isothermal amplification. Preprint at medRxiv https://doi.org/10.1101/2020.02.19.20025155 (2020).
Ding, Y., Chan, C. Y. & Lawrence, C. E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 32, W135–W141 (2004).
Pacchioni, G. A not-so-strong bond. Nat. Rev. Mater. 4, 226–226 (2019).
Abdulhay, E., Alafeef, M., Hadoush, H. & Arunkumar, N. Resting state EEG-based diagnosis of Autism via elliptic area of continuous wavelet transform complex plot. Preprint in J. Intell. Fuzzy Syst. https://doi.org/10.3233/JIFS-189176 (2020).
Abdulhay, E., Alafeef, M., Abdelhay, A. & Al-Bashir, A. Classification of normal, ictal and inter-ictal EEG via direct quadrature and random forest tree. J. Med. Biol. Eng. 37, 843–857 (2017).
Hadoush, H., Nazzal, M., Almasri, N. A., Khalil, H. & Alafeef, M. Therapeutic effects of bilateral anodal transcranial direct current stimulation on prefrontal and motor cortical areas in children with autism spectrum disorders: a pilot study. Autism Res. 13, 828–836 (2020).
Alafeef, M. Smartphone-based photoplethysmographic imaging for heart rate monitoring. J. Med. Eng. Technol. 41, 387–395 (2017).
Alafeef, M. & Fraiwan, M. Smartphone-based respiratory rate estimation using photoplethysmographic imaging and discrete wavelet transform. J. Ambient Intell. Humaniz. Comput. 11, 693–703 (2020).
Alafeef, M., Fraiwan, M., Alkhalaf, H. & Audat, Z. Shannon entropy and fuzzy C-means weighting for AI-based diagnosis of vertebral column diseases. J. Ambient Intell. Humaniz. Comput. 11, 2557–2566 (2020).
Rump, L. V., Asamoah, B. & Gonzalez-Escalona, N. Comparison of commercial RNA extraction kits for preparation of DNA-free total RNA from Salmonella cells. BMC Res. Notes 3, 211 (2010).
Kamra, M., Moitra, P., Ponnalagu, D., Karande, A. A. & Bhattacharya, S. New water-soluble oxyamino chitosans as biocompatible vectors for efficacious anticancer therapy via co-delivery of gene and drug. ACS Appl. Mater. Interfaces 11, 37442–37460 (2019).
Teubl, B. J. et al. The effect of saliva on the fate of nanoparticles. Clin. Oral Invest. 22, 929–940 (2018).
Ngamchuea, K., Batchelor-McAuley, C. & Compton, R. G. The fate of silver nanoparticles in authentic human saliva. Nanotoxicology 12, 305–311 (2018).
Acknowledgements
The authors gratefully acknowledge the receipt of funding from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) R03EB028026, R03 EB028026-02S2 and R03 EB028026-02S1, the University of Maryland, Baltimore (UMB) and the University of Maryland, Baltimore County (UMBC). The authors thank Prof. Z. Ahmed’s Lab from the Department of Otorhinolaryngology-Head and Neck Surgery, UMB, for providing access to the PCR machine. The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: (i) SARS-related coronavirus 2, isolate USA-WA1/2020, gamma-irradiated, NR-52287, and (ii) the MERS CoV, EMC/2012, irradiated infected cell lysate, NR-50549. Figures 1–4 were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
M.A., P.M., K.D. and D.P. conceived the project, designed the experiments and wrote the manuscript. D.P. provided study direction. M.A. and P.M. designed the primers and the ASOs. M.A., P.M. and K.D. tested the kit performance using clinical and spiked samples and performed the experiments. All the authors tested the protocol and provided feedback. All authors have read and approved the final version of the manuscript. M.A., P.M. and K.D. contributed equally to this study.
Corresponding author
Ethics declarations
Competing interests
D.P. is the founder or cofounder of three university-based start-ups. None of these entities, however, supported this work. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Protocols thanks Elba Mauriz, John Zhang and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Moitra, P. et al. ACS Nano 14, 7617–7627 (2020): https://doi.org/10.1021/acsnano.0c03822
Alafeef, M. et al. ACS Nano 14, 17028–17045 (2020): https://doi.org/10.1021/acsnano.0c06392
Moitra, P. et al. Preprint at medRxiv (2020): https://doi.org/10.1101/2020.10.08.20208785
Supplementary information
Supplementary Information
Supplementary Discussion, Data 1, Figs. 1–4, Tables 1–5 and References.
Source data
Rights and permissions
About this article

Cite this article
Alafeef, M., Moitra, P., Dighe, K. et al. RNA-extraction-free nano-amplified colorimetric test for point-of-care clinical diagnosis of COVID-19. Nat Protoc 16, 3141–3162 (2021). https://doi.org/10.1038/s41596-021-00546-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-021-00546-w
This article is cited by
-
Nanoplasmonic amplification in microfluidics enables accelerated colorimetric quantification of nucleic acid biomarkers from pathogens
Nature Nanotechnology (2023)
-
Vivid COVID-19 LAMP is an ultrasensitive, quadruplexed test using LNA-modified primers and a zinc ion and 5-Br-PAPS colorimetric detection system
Communications Biology (2023)
-
AuNP-based biosensors for the diagnosis of pathogenic human coronaviruses: COVID-19 pandemic developments
Analytical and Bioanalytical Chemistry (2022)
-
A rapid near-patient detection system for SARS-CoV-2 using saliva
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
