
Abstract
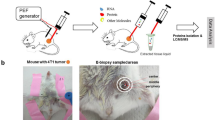
Bottom-up mass spectrometry–based proteomics relies on protein digestion and peptide purification. The application of such methods to broadly available clinical samples such as formalin-fixed and paraffin-embedded (FFPE) tissues requires reversal of chemical crosslinking and the removal of reagents that are incompatible with mass spectrometry. Here, we describe in detail a protocol that combines tissue disruption by ultrasonication, heat-induced antigen retrieval and two alternative methods for efficient detergent removal to enable quantitative proteomic analysis of limited amounts of FFPE material. To show the applicability of our approach, we used hepatocellular carcinoma (HCC) as a model system. By combining the described protocol with laser-capture microdissection, we were able to quantify the intra-tumor heterogeneity of a tumor specimen on the proteome level using a single slide with tissue of 10-µm thickness. We also demonstrate broader applicability to other tissues, including human gallbladder and heart. The procedure described in this protocol can be completed within 8 d.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
TMT and DIA data of the HCC samples shown in Figs. 4 and 5 are available via the ProteomeXchange Consortium (http://www.proteomexchange.org/) or the PRIDE Proteomics Identification Database (https://www.ebi.ac.uk/pride/) with the dataset identifier PXD007052. Lists of proteins identified in the experiments shown in Fig. 4 are provided in Supplementary Table 1. Raw data for gallbladder (Fig. 4e) and heart (Fig. 4f) samples are available from the authors upon request.
References
Doll, S., Gnad, F. & Mann, M. The case for proteomics and phospho-proteomics in personalized cancer medicine. Proteom. Clin. Appl 13, 1–10 (2019).
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Drummond, E. S., Nayak, S., Ueberheide, B. & Wisniewski, T. Proteomic analysis of neurons microdissected from formalin-fixed, paraffin-embedded Alzheimer’s disease brain tissue. Sci. Rep. 5, 15456 (2015).
Liu, A. Laser capture microdissection in the tissue biorepository. J. Biomol. Tech. 21, 120–125 (2010).
Ostasiewicz, P., Zielinska, D. F., Mann, M. & Wiśniewski, J. R. Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J. Proteome Res. 9, 3688–3700 (2010).
Wiśniewski, J. R., Ostasiewicz, P. & Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 10, 3040–3049 (2011).
Karlsson, C. & Karlsson, M. G. Effects of long-term storage on the detection of proteins, DNA, and mRNA in tissue microarray slides. J. Histochem. Cytochem. 59, 1113–1121 (2011).
Föll, M. C. et al. Reproducible proteomics sample preparation for single FFPE tissue slices using acid-labile surfactant and direct trypsinization. Clin. Proteom. 15, 11 (2018).
Bayer, M., Angenendt, L., Schliemann, C., Hartmann, W. & König, S. Are formalin-fixed and paraffin-embedded tissues fit for proteomic analysis? J. Mass Spectrom. https://doi.org/10.1002/jms.4347 (2019).
Longuespée, R. et al. A laser microdissection-based workflow for FFPE tissue microproteomics: important considerations for small sample processing. Methods 104, 154–162 (2016).
Marakalala, M. J. et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat. Med 22, 531–538 (2016).
Davis, S., Scott, C., Ansorge, O. & Fischer, R. Development of a sensitive, scalable method for spatial, cell-type-resolved proteomics of the human brain. J. Proteome Res. 18, 1787–1795 (2019).
Guo, T. et al. Multi-region proteome analysis quantifies spatial heterogeneity of prostate tissue biomarkers. Life Sci. Alliance 1, e201800042 (2018).
Buczak, K. et al. Spatial tissue proteomics quantifies inter- and intratumor heterogeneity in hepatocellular carcinoma (HCC). Mol. Cell. Proteom. 17, 810–825 (2018).
Wiśniewski, J. R. et al. Absolute proteome analysis of colorectal mucosa, adenoma, and cancer reveals drastic changes in fatty acid metabolism and plasma membrane transporters. J. Proteome Res. 14, 4005–4018 (2015).
Shi, S. R., Key, M. E. & Kalra, K. L. Antigen retrieval in formalin-fixed, paraffin-embedded tissues: an enhancement method for immunohistochemical staining based on microwave oven heating of tissue sections. J. Histochem. Cytochem. 39, 741–748 (1991).
Hughes, C. S. et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 10, 757 (2014).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 14, 68–85 (2019).
Sielaff, M. et al. Evaluation of FASP, SP3, and iST Protocols for proteomic sample preparation in the low microgram range. J. Proteome Res. 16, 4060–4072 (2017).
Hughes, C. S. et al. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Sci. Rep. 6, 34949 (2016).
Müller, T. et al. Automated sample preparation with SP 3 for low‐input clinical proteomics. Mol. Syst. Biol. 16, 1–19 (2020).
Heinze, I. et al. Species comparison of liver proteomes reveals links to naked mole-rat longevity and human aging. BMC Biol. 16, 82 (2018).
Thompson, A. et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 (2003).
Gillet, L. C. et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteom. 11, O111.016717 (2012).
Jackson, H. W. et al. The single-cell pathology landscape of breast cancer. Nature 578, 615–620 (2020).
Ijsselsteijn, M. E., van der Breggen, R., Farina Sarasqueta, A., Koning, F. & de Miranda, N. F. C. C. A 40-marker panel for high dimensional characterization of cancer immune microenvironments by imaging mass cytometry. Front. Immunol. 10, 1–8 (2019).
Judd, A. M. et al. A recommended and verified procedure for in situ tryptic digestion of formalin-fixed paraffin-embedded tissues for analysis by matrix-assisted laser desorption/ionization imaging mass spectrometry. J. Mass Spectrom. 54, 716–727 (2019).
Li, J. et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 17, 399–404 (2020).
Muntel, J. et al. Comparison of Protein quantification in a complex background by DIA and TMT workflows with fixed instrument time. J. Proteome Res. 18, 1340–1351 (2019).
Bekker-Jensen, D. B. et al. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 11, 1–12 (2020).
Huang, T. et al. Combining precursor and fragment information for improved detection of differential abundance in data independent acquisition. Mol. Cell. Proteom. 19, 421–430 (2020).
Butler, S. L. et al. The antigen for Hep Par 1 antibody is the urea cycle enzyme carbamoyl phosphate synthetase 1. Lab. Invest. 88, 78–88 (2008).
Muntel, J. et al. Surpassing 10 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omi 15, 348–360 (2019).
Skillbäck, T. et al. A novel quantification-driven proteomic strategy identifies an endogenous peptide of pleiotrophin as a new biomarker of Alzheimer’s disease. Sci. Rep. 7, 1–12 (2017).
Brenes, A., Hukelmann, J., Bensaddek, D. & Lamond, A. I. Multibatch TMT reveals false positives, batch effects and missing values. Mol. Cell. Proteom. 18, 1967–1980 (2019).
Bruderer, R. et al. Analysis of 1508 plasma samples by capillary-flow data-independent acquisition profiles proteomics of weight loss and maintenance. Mol. Cell. Proteom. 18, 1242–1254 (2019).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Tsou, C. C. et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264 (2015).
Röst, H. L. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014).
Pino, L. K. et al. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 39, 229–244 (2020).
Viswanadhapalli, S. et al. Estrogen receptor coregulator binding modulator (ERX-11) enhances the activity of CDK4/6 inhibitors against estrogen receptor-positive breast cancers. Breast Cancer Res 21, 150 (2019).
Tran, N. H. et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat. Methods 16, 63–66 (2019).
Choi, M. et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014).
Gatto, L. & Lilley, K. S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28, 288–289 (2011).
Acknowledgements
The authors acknowledge I. Heinze and the CF Proteomics of the FLI for technical support; members of the tissue bank of the National Center for Tumor disease (NCT) Heidelberg, in particular E. Herpel and V. Geissler, for their support; as well as J. Scheuerer for her support with the laser microdissection; and L. Reiter and J. Muntel for critical reading of the manuscript. M.B. acknowledges funding from the European Molecular Biology Laboratory and the Max Planck Society. L.F. acknowledges financial support from project ERAatUC, grant no. 669088, under the Horizon 2020 program of the European Commission. D.S. was supported by a PhD fellowship from the Portuguese Foundation for Science and Technology (FCT, PD/BD/106051/2015) under the Inter-University Doctoral Program in Aging and Chronic Diseases. S.R. acknowledges funding from the Wilhelm Sander-Stiftung (no. 2015.111.1) and was supported in part by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; Project ID 314905040 – TRR 209 within Project B01). S.S. acknowledges funding from the DFG: SFB/TR209 (B04) and Si 1487/3-1, from the Hella-Buehler-Foundation and from an HRCMM (Heidelberg Research Center for Molecular Medicine) Career Development Fellowship. A.O. acknowledges funding from DFG via the Research Training Group ProMoAge (GRK 2155), the Else Kröner Fresenius Stiftung (award no. 2019_A79) and the Deutsches Zentrum für Herz-Kreislaufforschung (award no. 81X2800193). The FLI is a member of the Leibniz Association and is financially supported by the Federal Government of Germany and the State of Thuringia.
Author information
Authors and Affiliations
Contributions
Conceptualization: K.B., J.M.K., S.S., M.B., A.O. Data analysis: K.B., J.M.K., A.O. Investigation: K.B., J.M.K., F.T., D.S. Methodology: K.B., J.M.K., S.S., M.B., A.O. Project administration: M.B., A.O. Data analysis: K.B., J.M.K., A.O. Supervision: S.R., L.F., M.B., A.O. Visualization: K.B., A.O. Writing (original draft): K.B., J.M.K., M.B., A.O. Writing (review and editing): S.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Buczak, K. et al. Mol. Cell. Proteomics 17, 810–825 (2018): https://www.mcponline.org/content/17/4/810.long
Heinze, I. et al. BMC Biol. 16, 82 (2018): https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-018-0547-y
Supplementary information
Supplementary Table 1
Proteins identified in the experiments shown in Fig. 4.
Rights and permissions
About this article

Cite this article
Buczak, K., Kirkpatrick, J.M., Truckenmueller, F. et al. Spatially resolved analysis of FFPE tissue proteomes by quantitative mass spectrometry. Nat Protoc 15, 2956–2979 (2020). https://doi.org/10.1038/s41596-020-0356-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-020-0356-y
This article is cited by
-
Mass spectrometry-based proteomics as an emerging tool in clinical laboratories
Clinical Proteomics (2023)
-
A distinct circular DNA profile intersects with proteome changes in the genotoxic stress-related hSOD1G93A model of ALS
Cell & Bioscience (2023)
-
Multi-omics analysis identifies RFX7 targets involved in tumor suppression and neuronal processes
Cell Death Discovery (2023)
-
Proteomics to study cancer immunity and improve treatment
Seminars in Immunopathology (2023)
-
Spatial mapping of cellular senescence: emerging challenges and opportunities
Nature Aging (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
