
Abstract
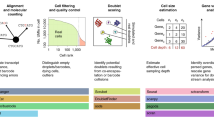
Single-cell RNA sequencing (scRNA-seq) is a popular and powerful technology that allows you to profile the whole transcriptome of a large number of individual cells. However, the analysis of the large volumes of data generated from these experiments requires specialized statistical and computational methods. Here we present an overview of the computational workflow involved in processing scRNA-seq data. We discuss some of the most common tasks and the tools available for addressing central biological questions. In this article and our companion website (https://scrnaseq-course.cog.sanger.ac.uk/website/index.html), we provide guidelines regarding best practices for performing computational analyses. This tutorial provides a hands-on guide for experimentalists interested in analyzing their data as well as an overview for bioinformaticians seeking to develop new computational methods.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

References
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Svensson, V. et al. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 14, 381–387 (2017).
Ziegenhain, C. et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643 (2017).
Han, X. et al. Mapping the mouse cell atlas by microwell-seq. Cell 172, 1091–1107 (2018).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele- and isoform-resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Zhang, X. et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell 73, 130–142 (2019).
Wu, A. R. et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 11, 41–46 (2014).
Sarkar, A. K. & Stephens, M. Separating measurement and expression models clarifies confusion in single cell RNA-seq analysis. Preprint at https://www.biorxiv.org/content/10.1101/2020.04.07.030007v1 (2020).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Lun, A. T. L. et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
Amezquita, R. A. et al. Orchestrating single-cell analysis with bioconductor. Nat. Methods 17, 137–145 (2020).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291 (2019).
Lareau, C. A., Ma, S., Duarte, F. M. & Buenrostro, J. D. Inference and effects of barcode multiplets in droplet-based single-cell assays. Nat. Commun. 11, 866 (2020).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337 (2019).
Bais, A. S. & Kostka, D. scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics 36, 1150–1158 (2020).
Marinov, G. K. et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 24, 496–510 (2014).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Lun, A. T. L., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016).
Lun, A. T. L., Calero-Nieto, F. J., Haim-Vilmovsky, L., Göttgens, B. & Marioni, J. C. Assessing the reliability of spike-in normalization for analyses of single-cell RNA sequencing data. Genome Res. 27, 1795–1806 (2017).
Bacher, R. et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584–586 (2017).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Tang, W. et al. bayNorm: Bayesian gene expression recovery, imputation and normalization for single-cell RNA-sequencing data. Bioinformatics 36, 1174–1181 (2020).
Baran-Gale, J., Chandra, T. & Kirschner, K. Experimental design for single-cell RNA sequencing. Brief Funct. Genomics 17, 233–239 (2018).
Stein, C. K. et al. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat. BMC Bioinformatics 16, 63 (2015).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Li, W. V. & Li, J. J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 9, 997 (2018).
Gong, W., Kwak, I.-Y., Pota, P., Koyano-Nakagawa, N. & Garry, D. J. DrImpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinformatics 19, 220 (2018).
Huang, M. et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542 (2018).
Andrews, T. S. & Hemberg, M. False signals induced by single-cell imputation. F1000Research 7, 1740 (2018).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Wang, J. et al. Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods 16, 875–878 (2019).
Elyanow, R., Dumitrascu, B., Engelhardt, B. E. & Raphael, B. J. netNMF-sc: leveraging gene–gene interactions for imputation and dimensionality reduction in single-cell expression analysis. Genome Res. 30, 195–204 (2020).
Scialdone, A. et al. Computational assignment of cell-cycle stage from single-cell transcriptome data. Methods 85, 54–61 (2015).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Andrews, T. S. & Hemberg, M. M3Drop: dropout-based feature selection for scRNASeq. Bioinformatics 35, 2865–2867 (2019).
Yip, S. H., Sham, P. C. & Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief Bioinformatics 20, 1583–1589 (2019).
Svensson, V. Droplet scRNA-seq is not zero-inflated. Nat. Biotechnol. 38, 147–150 (2020).
Jiang, L., Chen, H., Pinello, L. & Yuan, G.-C. GiniClust: detecting rare cell types from single-cell gene expression data with Gini index. Genome Biol. 17, 144 (2016).
Sun, S., Zhu, J., Ma, Y. & Zhou, X. Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis. Genome Biol. 20, 269 (2019).
Peres-Neto, P. R., Jackson, D. A. & Somers, K. M. How many principal components? stopping rules for determining the number of non-trivial axes revisited. Comput. Stat. Data Anal. 49, 974–997 (2005).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Software 3, 861 (2018).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Kobak, D. & Linderman, G. C. UMAP does not preserve global structure any better than t-SNE when using the same initialization. Preprint at https://www.biorxiv.org/content/10.1101/2019.12.19.877522v1 (2019).
Hartigan, J. A. & Wong, M. A. Algorithm AS 136: a k-means clustering algorithm. Appl. Stat. 28, 100–108 (1979).
Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Levine, J. H. et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Research 7, 1141 (2018).
Freytag, S., Tian, L., Lönnstedt, I., Ng, M. & Bahlo, M. Comparison of clustering tools in R for medium-sized 10× Genomics single-cell RNA-sequencing data. F1000Research 7, 1297 (2018).
Kiselev, V. Y., Andrews, T. S. & Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 20, 273–282 (2019).
Zappia, L. & Oshlack, A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. Gigascience 7, giy083 (2018).
Cannoodt, R., Saelens, W. & Saeys, Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur. J. Immunol. 46, 2496–2506 (2016).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Ji, Z. & Ji, H. TSCAN: pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 44, e117 (2016).
Chen, J., Schlitzer, A., Chakarov, S., Ginhoux, F. & Poidinger, M. Mpath maps multi-branching single-cell trajectories revealing progenitor cell progression during development. Nat. Commun. 7, 11988 (2016).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-0591-3 (2020).
Zhang, J. M., Kamath, G. M. & Tse, D. N. Valid post-clustering differential analysis for single-cell RNA-seq. Cell Syst. 9, 383–392 (2019).
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
Van den Berge, K. et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 19, 24 (2018).
Vieth, B., Parekh, S., Ziegenhain, C., Enard, W. & Hellmann, I. A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 10, 4667 (2019).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Crowell, H. L. et al. On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data. Preprint at https://www.biorxiv.org/content/10.1101/713412v1 (2019).
Baran, Y. et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019).
Kiselev, V. Y., Yiu, A. & Hemberg, M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2018).
Abdelaal, T. et al. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 20, 194 (2019).
Crow, M., Paul, A., Ballouz, S., Huang, Z. J. & Gillis, J. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun. 9, 884 (2018).
Macaulay, I. C., Ponting, C. P. & Voet, T. Single-cell multiomics: multiple measurements from single cells. Trends Genet. 33, 155–168 (2017).
Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272 (2019).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
Brunet Avalos, C., Maier, G. L., Bruggmann, R. & Sprecher, S. G. Single cell transcriptome atlas of the Drosophila larval brain. eLife 8, e50354 (2019).
Tabula Muris Consortium. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
McCarthy, D. J., Campbell, K. R., Lun, A. T. L. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Pliner, H. A., Shendure, J. & Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 16, 983–986 (2019).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Acknowledgements
We thank J. Eliasova for help with Fig. 1. We also thank S. Ballerau, M. Büttner, M. Do Nascimentoo Lopes Primo, J. Lee, R. Lyu, E. Madissoon, R. Martinez Nunez, S. Y. Müller, K. Polanski, P. Qiao and J. Westoby for their contributions to teaching the course and developing the material.
Author information
Authors and Affiliations
Contributions
T.S.A., V.Y.K., D.M. and M.H. planned the tutorial and wrote the text together.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Andrews, T.S., Kiselev, V.Y., McCarthy, D. et al. Tutorial: guidelines for the computational analysis of single-cell RNA sequencing data. Nat Protoc 16, 1–9 (2021). https://doi.org/10.1038/s41596-020-00409-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-020-00409-w
This article is cited by
-
The implications of single-cell RNA-seq analysis in prostate cancer: unraveling tumor heterogeneity, therapeutic implications and pathways towards personalized therapy
Military Medical Research (2024)
-
Cellograph: a semi-supervised approach to analyzing multi-condition single-cell RNA-sequencing data using graph neural networks
BMC Bioinformatics (2024)
-
Multimodal Nature of the Single-cell Primate Brain Atlas: Morphology, Transcriptome, Electrophysiology, and Connectivity
Neuroscience Bulletin (2024)
-
Meningeal immunity and neurological diseases: new approaches, new insights
Journal of Neuroinflammation (2023)
-
scRNASequest: an ecosystem of scRNA-seq analysis, visualization, and publishing
BMC Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
