
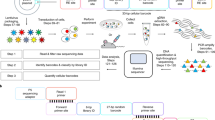
Abstract
Single-cell technologies are offering unparalleled insight into complex biology, revealing the behavior of rare cell populations that are masked in bulk population analyses. One current limitation of single-cell approaches is that lineage relationships are typically lost as a result of cell processing. We recently established a method, CellTagging, permitting the parallel capture of lineage information and cell identity via a combinatorial cell indexing approach. CellTagging integrates with high-throughput single-cell RNA sequencing, where sequential rounds of cell labeling enable the construction of multi-level lineage trees. Here, we provide a detailed protocol to (i) generate complex plasmid and lentivirus CellTag libraries for labeling of cells; (ii) sequentially CellTag cells over the course of a biological process; (iii) profile single-cell transcriptomes via high-throughput droplet-based platforms; and (iv) generate a CellTag expression matrix, followed by clone calling and lineage reconstruction. This lentiviral-labeling approach can be deployed in any organism or in vitro culture system that is amenable to viral transduction to simultaneously profile lineage and identity at single-cell resolution.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout










Similar content being viewed by others


Data availability
CellTagging of fibroblast to iEP lineage reprogramming42 data are available via GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99915. The clones and lineages reconstructed from this dataset can be interactively explored via http://celltag.org/, along with our simulator to support CellTag experimental design. CellTagging constructs are available from Addgene: https://www.addgene.org/pooled-library/morris-lab-celltag/. Updates to this protocol will be provided at https://www.protocols.io/view/single-cell-mapping-of-lineage-and-identity-via-ce-yxifxke.
Code availability
Our R package, CellTagR, code and analysis tutorials are available via GitHub: https://github.com/morris-lab/CellTagR.
References
Regev, A. et al. The human cell atlas. Elife 6, 27041 (2017).
Han, X. et al. Mapping the mouse cell atlas by microwell-seq. Cell 172, 1091–1107.e17 (2018).
Tabula Muris Consortium. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Ramsköld, D. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777–782 (2012).
Streets, A. M. et al. Microfluidic single-cell whole-transcriptome sequencing. Proc. Natl Acad. Sci. USA 111, 7048–7053 (2014).
Macosko, E. Z. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Klein, A. M. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Cao, J. et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017).
Cusanovich, D. A. et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324 (2018).
Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019).
Cao, J. et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018).
Welch, J. D. et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177, 1873–1887.e17 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Guo, M., Bao, E. L., Wagner, M., Whitsett, J. A. & Xu, Y. SLICE: determining cell differentiation and lineage based on single cell entropy. Nucleic Acids Res. 45, e54 (2016).
Ji, Z. & Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 44, e117 (2016).
Shin, J. et al. Single-cell RNA-seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell 17, 360–372 (2015).
Marco, E. et al. Bifurcation analysis of single-cell gene expression data reveals epigenetic landscape. Proc. Natl Acad. Sci. USA 111, 5643–5650 (2014).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Setty, M. et al. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 34, 637–645 (2016).
Bendall, S. C. et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Grün, D. et al. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 19, 266–277 (2016).
Chen, J., Schlitzer, A., Chakarov, S., Ginhoux, F. & Poidinger, M. Mpath maps multi-branching single-cell trajectories revealing progenitor cell progression during development. Nat. Commun. 7, 11988 (2016).
Weinreb, C., Wolock, S., Tusi, B. K., Socolovsky, M. & Klein, A. M. Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl Acad. Sci. Usa. 115, E2467–E2476 (2018).
Giecold, G., Marco, E., Garcia, S. P., Trippa, L. & Yuan, G.-C. Robust lineage reconstruction from high-dimensional single-cell data. Nucleic Acids Res. 44, e122 (2016).
Lodato, M. A. et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015).
Leung, M. L. et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 27, 1287–1299 (2017).
Ludwig, L. S. et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339.e22 (2019).
Kester, L. & van Oudenaarden, A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 23, 166–179 (2018).
Lu, R., Neff, N. F., Quake, S. R. & Weissman, I. L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat. Biotechnol. 29, 928–933 (2011).
Porter, S. N., Baker, L. C., Mittelman, D. & Porteus, M. H. Lentiviral and targeted cellular barcoding reveals ongoing clonal dynamics of cell lines in vitro and in vivo. Genome Biol. 15, R75 (2014).
Sun, J. et al. Clonal dynamics of native haematopoiesis. Nature 514, 322–327 (2014).
McKenna, A. et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016).
Frieda, K. L. et al. Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017).
Schmidt, S. T., Zimmerman, S. M., Wang, J., Kim, S. K. & Quake, S. R. Quantitative analysis of synthetic cell lineage tracing using nuclease barcoding. ACS Synth. Biol. 6, 936–942 (2017).
Biddy, B. A. et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224 (2018).
Yao, Z. et al. A single-cell roadmap of lineage bifurcation in human ESC models of embryonic brain development. Cell Stem Cell 20, 120–134 (2017).
Spanjaard, B. et al. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol. 36, 469–473 (2018).
Raj, B. et al. Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol. 36, 442–450 (2018).
Alemany, A., Florescu, M., Baron, C. S., Peterson-Maduro, J. & van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018).
Wagner, D. E. et al. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987 (2018).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science eaaw3381 (2020).
Guo, C. et al. CellTag indexing: genetic barcode-based sample multiplexing for single-cell genomics. Genome Biol. 20, 90 (2019).
van Galen, P. et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–72 (2014).
Turner, D. L. & Cepko, C. L. A common progenitor for neurons and glia persists in rat retina late in development. Nature 328, 131–136 (1987).
Frank, E. & Sanes, J. R. Lineage of neurons and glia in chick dorsal root ganglia: analysis in vivo with a recombinant retrovirus. Development 111, 895–908 (1991).
Pei, W. et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 548, 456–460 (2017).
Chan, M. M. et al. Molecular recording of mammalian embryogenesis. Nature 1, 77–82 (2019).
Kalhor, R. et al. Developmental barcoding of whole mouse via homing CRISPR. Science 361, eaat9804 (2018).
Raj, B., Gagnon, J. A. & Schier, A. F. Large-scale reconstruction of cell lineages using single-cell readout of transcriptomes and CRISPR–Cas9 barcodes by scGESTALT. Nat. Protoc. 13, 2685–2713 (2018).
Kalhor, R., Mali, P. & Church, G. M. Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017).
Perli, S. D., Cui, C. H. & Lu, T. K. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353, aag0511 (2016).
Morris, S. A. et al. Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell 158, 889–902 (2014).
Doulatov, S. et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell 13, 459–470 (2013).
Kita-Matsuo, H. et al. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS ONE 4, e5046 (2009).
Hong, S. et al. Functional analysis of various promoters in lentiviral vectors at different stages of in vitro differentiation of mouse embryonic stem cells. Mol. Ther. 15, 1630–1639 (2007).
Ramezani, A. & Hawley, R. G. Strategies to insulate lentiviral vector-expressed transgenes. Methods Mol. Biol. 614, 77–100 (2010).
Benabdellah, K., Gutierrez-Guerrero, A., Cobo, M., Muñoz, P. & Martín, F. A chimeric HS4-SAR insulator (IS2) that prevents silencing and enhances expression of lentiviral vectors in pluripotent stem cells. PLoS ONE 9, e84268 (2014).
Pfaff, N. et al. A ubiquitous chromatin opening element prevents transgene silencing in pluripotent stem cells and their differentiated progeny. Stem Cells 31, 488–499 (2013).
Alles, J. et al. Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biol. 15, 44 (2017).
Garfield, A. S. Derivation of primary mouse embryonic fibroblast (PMEF) cultures. Methods Mol. Biol. 633, 19–27 (2010).
Ichim, C. V. & Wells, R. A. Generation of high-titer viral preparations by concentration using successive rounds of ultracentrifugation. J. Transl. Med. 9, 137 (2011).
Zorita, E., Cuscó, P. & Filion, G. J. Starcode: sequence clustering based on all-pairs search. Bioinformatics 31, 1913–1919 (2015).
Berggren, W. T., Lutz, M. & Modesto, V. General spinfection protocol. in StemBook (ed The Stem Cell Community) (Harvard Stem Cell Institute, 2008).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2018).
Acknowledgements
We thank members of the Morris laboratory for critical discussions and J. Dick (University of Toronto) for the gift of the pSMAL backbone. This work was funded by National Institutes of Health grants R01-GM126112, R21-HG009750 and P30-DK052574; Silicon Valley Community Foundation, Chan Zuckerberg Initiative Grants HCA-A-1704-01646 and HCA2-A-1708-02799; The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital grant MI-II-2016-544. S.A.M. is supported by a Vallee Scholar Award; B.A.B. is supported by NIH-T32HG000045-18; and K.K. is supported by a Japan Society for the Promotion of Science Postdoctoral Fellowship.
Author information
Authors and Affiliations
Contributions
S.A.M., W.K. and B.A.B. developed and optimized the CellTagging protocols and analyzed the data. K.K. developed CellTag lineage tree reconstuction. J.M.A. and E.G.B. developed the CellTag simulator. W.K. and S.A.M. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Protocols thanks Jennifer Adair, James Gagnon and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Biddy, B. A. et al. Nature 564, 219–224 (2018): https://doi.org/10.1038/s41586-018-0744-4
Guo, C. et al. Genome Biol. 20, 90 (2019): https://doi.org/10.1186/s13059-019-1699-y
Supplementary information
Supplementary Manual 1
A step-by-step tutorial for CellTag data processing using the CellTagR package.
Supplementary Software 1
12 scripts comprising the CellTagR package
Supplementary Software 2
Script for the CellTag simulation tool
Rights and permissions
About this article

Cite this article
Kong, W., Biddy, B.A., Kamimoto, K. et al. CellTagging: combinatorial indexing to simultaneously map lineage and identity at single-cell resolution. Nat Protoc 15, 750–772 (2020). https://doi.org/10.1038/s41596-019-0247-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-019-0247-2
This article is cited by
-
Extracting, filtering and simulating cellular barcodes using CellBarcode tools
Nature Computational Science (2024)
-
Combining single-cell transcriptomics and CellTagging to identify differentiation trajectories of human adipose-derived mesenchymal stem cells
Stem Cell Research & Therapy (2023)
-
Single-cell lineage capture across genomic modalities with CellTag-multi reveals fate-specific gene regulatory changes
Nature Biotechnology (2023)
-
Clonal tracking in cancer and metastasis
Cancer and Metastasis Reviews (2023)
-
Spliceosome inhibitor induces human hematopoietic progenitor cell reprogramming toward stemness
Experimental Hematology & Oncology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
