
Abstract
Chromatin immunoprecipitation coupled to next-generation sequencing (ChIP-seq) has served as the central method for the study of histone modifications for the past decade. In ChIP-seq analyses, antibodies selectively capture nucleosomes bearing a modification of interest and the associated DNA is then mapped to the genome to determine the distribution of the mark. This approach has several important drawbacks: (i) ChIP interpretation necessitates the assumption of perfect antibody specificity, despite growing evidence that this is often not the case. (ii) Common methods for evaluating antibody specificity in other formats have little or no bearing on specificity within a ChIP experiment. (iii) Uncalibrated ChIP is reported as relative enrichment, which is biologically meaningless outside the experimental reference frame defined by a discrete immunoprecipitation (IP), thus preventing facile comparison across experimental conditions or modifications. (iv) Differential library amplification and loading onto next-generation sequencers, as well as computational normalization, can further compromise quantitative relationships that may exist between samples. Consequently, the researcher is presented with a series of potential pitfalls and is blind to nearly all of them. Here we provide a detailed protocol for internally calibrated ChIP (ICeChIP), a method we recently developed to resolve these problems by spike-in of defined nucleosomal standards within a ChIP procedure. This protocol is optimized for specificity and quantitative power, allowing for measurement of antibody specificity and absolute measurement of histone modification density (HMD) at genomic loci on a biologically meaningful scale enabling unambiguous comparisons. We provide guidance on optimal conditions for next-generation sequencing (NGS) and instructions for data analysis. This protocol takes between 17 and 18 h, excluding time for sequencing or bioinformatic analysis. The ICeChIP procedure enables accurate measurement of histone post-translational modifications (PTMs) genome-wide in mammalian cells as well as Drosophila melanogaster and Caenorhabditis elegans, indicating suitability for use in eukaryotic cells more broadly.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

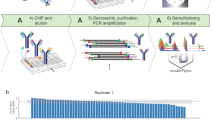
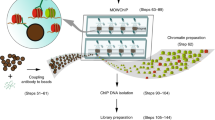
Code availability
All code used in this work is listed under Analytical tools. Scripts provided here are under GNU General Public License. The most updated versions of the tools and scripts described in this work can be found at http://github.com/shah-rohan/icechip.
References
Luger, K., Mäder, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Brand, M., Rampalli, S., Chaturvedi, C.-P. & Dilworth, F. J. Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc. 3, 398–409 (2008).
Jozwik, K. M., Chernukhin, I., Serandour, A. A., Nagarajan, S. & Carroll, J. S. FOXA1 directs H3K4 monomethylation at enhancers via recruitment of the methyltransferase MLL3. Cell Rep. 17, 2715–2723 (2016).
Rada-Iglesias, A. et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283 (2011).
Heintzman, N. D. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 (2007).
Heintzman, N. D. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112 (2009).
Calo, E. & Wysocka, J. Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 (2013).
Wang, C. et al. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc. Natl Acad. Sci. USA 113, 11871–11876 (2016).
Cheng, J. et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol. Cell 53, 979–992 (2014).
Wang, Y., Li, X. & Hu, H. H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228 (2014).
Fang, R. et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 39, 222–233 (2010).
Pekowska, A. et al. H3K4 tri‐methylation provides an epigenetic signature of active enhancers. EMBO J. 30, 4198–4210 (2011).
Popova, E. Y., Pinzon-Guzman, C., Salzberg, A. C., Zhang, S. S.-M. & Barnstable, C. J. LSD1-mediated demethylation of H3K4me2 is required for the transition from late progenitor to differentiated mouse rod photoreceptor. Mol. Neurobiol. 53, 4563–4581 (2016).
Zhang, J., Parvin, J. & Huang, K. Redistribution of H3K4me2 on neural tissue specific genes during mouse brain development. BMC Genomics 13, S5 (2012).
Barrero, M. J. et al. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 3, 1005–1011 (2013).
Bergmann, J. H. et al. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP‐A assembly on a synthetic human kinetochore. EMBO J. 30, 328–340 (2011).
Siklenka, K. et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 350, aab2006 (2015).
Santos-Rosa, H. et al. Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 (2002).
Schneider, J. et al. Molecular Regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Mol. Cell 19, 849–856 (2005).
Sims, R. J. III & Reinberg, D. Histone H3 Lys 4 methylation: caught in a bind? Genes Dev. 20, 2779–2786 (2006).
Sims, R. J. III et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 28, 665–676 (2007).
Ruthenburg, A. J., Allis, C. D. & Wysocka, J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell 25, 15–30 (2007).
Davie, J. R., Xu, W. & Delcuve, G. P. Histone H3K4 trimethylation: dynamic interplay with pre-mRNA splicing. Biochem. Cell Biol. 94, 1–11 (2015).
Shimazaki, N. & Lieber, M. R. Histone methylation and V(D)J recombination. Int. J. Hematol. 100, 230–237 (2014).
Vallianatos, C. N. & Iwase, S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics 7, 503–519 (2015).
Shen, E., Shulha, H., Weng, Z. & Akbarian, S. Regulation of histone H3K4 methylation in brain development and disease. Philos. Trans. R. Soc. B 369, 20130514 (2014).
Deb, M. et al. Chromatin dynamics: H3K4 methylation and H3 variant replacement during development and in cancer. Cell. Mol. Life Sci. 71, 3439–3463 (2014).
Gilmour, D. S. & Lis, J. T. Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc. Natl Acad. Sci. USA 81, 4275–4279 (1984).
Solomon, M. J. & Varshavsky, A. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc. Natl Acad. Sci. USA 82, 6470–6474 (1985).
Solomon, M. J., Larsen, P. L. & Varshavsky, A. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 53, 937–947 (1988).
Chen, H., Lin, R. J., Xie, W., Wilpitz, D. & Evans, R. M. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 98, 675–686 (1999).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Gifford, C. A. et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 153, 1149–1163 (2013).
Guenther, M. G. et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 22, 3403–3408 (2008).
Guenther, M. G., Levine, S. S., Boyer, L. A., Jaenisch, R. & Young, R. A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88 (2007).
Orlando, D. A. et al. Quantitative ChIP-seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170 (2014).
Xie, W. et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013).
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Grzybowski, A. T., Chen, Z. & Ruthenburg, A. J. Calibrating ChIP-seq with nucleosomal internal standards to measure histone modification density genome wide. Mol. Cell 58, 886–899 (2015).
Bonhoure, N. et al. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res. 24, 1157–1168 (2014).
Shah, R. N. et al. Examining the roles of H3K4 methylation states with systematically characterized antibodies. Mol. Cell 72, 162–177 (2018).
Bock, I. et al. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 6, 256–263 (2011).
Egelhofer, T. A. et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 18, 91–93 (2011).
Fuchs, S. M., Krajewski, K., Baker, R. W., Miller, V. L. & Strahl, B. D. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr. Biol. 21, 53–58 (2011).
Nishikori, S. et al. Broad ranges of affinity and specificity of anti-histone antibodies revealed by a quantitative peptide immunoprecipitation assay. J. Mol. Biol. 424, 391–399 (2012).
Hattori, T. et al. Recombinant antibodies to histone post-translational modifications. Nat. Methods 10, 992–995 (2013).
Rothbart, S. B. et al. An interactive database for the assessment of histone antibody specificity. Mol. Cell 59, 502–511 (2015).
Lowary, P. T. & Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276, 19–42 (1998).
Baker, M. Reproducibility crisis: Blame it on the antibodies. Nature 521, 274–276 (2015).
Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 533, 452–454 (2016).
Harris, R. Rigor Mortis: How Sloppy Science Creates Worthless Cures, Crushes Hopes, and Wastes Billions (Basic Books, 2017).
Guertin, M. J., Cullen, A. E., Markowetz, F. & Holding, A. N. Parallel factor ChIP provides essential internal control for quantitative differential ChIP-seq. Nucleic Acids Res. 46, e75 (2018).
Egan, B. et al. An alternative approach to ChIP-seq normalization enables detection of genome-wide changes in histone H3 lysine 27 trimethylation upon EZH2 inhibition. PLoS ONE 11, e0166438 (2016).
Lu, C. et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849 (2016).
Kasinathan, S., Orsi, G. A., Zentner, G. E., Ahmad, K. & Henikoff, S. High-resolution mapping of transcription factor binding sites on native chromatin. Nat. Methods 11, 203–209 (2014).
Teytelman, L. et al. Impact of chromatin structures on dna processing for genomic analyses. PLoS ONE 4, e6700 (2009).
Teytelman, L., Thurtle, D. M., Rine, J. & van Oudenaarden, A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl Acad. Sci. USA 110, 18602–18607 (2013).
Fan, X. & Struhl, K. Where does mediator bind in vivo? PLoS ONE 4, e5029 (2009).
Peng, Q., Vijaya Satya, R., Lewis, M., Randad, P. & Wang, Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics 16, 589 (2015).
LeRoy, G. et al. A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20 (2013).
Rohland, N. & Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 21, A.3B.1–A.3B.2 (1997).
Acknowledgements
This work was funded by the National Institutes of Health under award number R01-GM115945 to A.J.R.
Author information
Authors and Affiliations
Contributions
A.T.G. and A.J.R. conceived and developed ICeChIP. A.T.G. and R.N.S. wrote computational scripts for data analysis while A.J.R. provided oversight. W.F.R. independently conducted ICeChIP–seq analyses to validate this protocol. R.N.S. wrote the manuscript with input from the other authors.
Corresponding author
Ethics declarations
Competing interests
A.J.R. and A.T.G. hold partial intellectual property rights to ICeChIP as inventors. R.N.S., A.T.G. and A.J.R. have previously served in a compensated consulting role to Epicypher, the commercial developer and supplier of ICeChIP barcoded nucleosomes (SNAP-ChIP and CAP-ChIP, both of which are under a license of the University of Chicago (patent #US20160341743)).
Additional information
Peer review information Nature Protocols thanks Jeff Dilworth, Alon Goren, Yuting Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
Grzybowski, A. T., Chen, Z. & Ruthenburg, A. J. Mol. Cell 58, 886–899 (2015): https://doi.org/10.1016/j.molcel.2015.04.022
Werner, M. S. et al. Nat. Struct. Mol. Biol. 24, 596–603 (2017): https://doi.org/10.1038/nsmb.3424
Shah, R. N. et al. Mol. Cell 72, 162–177 (2018): https://doi.org/10.1016/j.molcel.2018.08.015
Supplementary information
Supplementary Table 1
Primer and probe sequences
Supplementary Table 2
Example of calibration table formatting
Supplementary Table 3
Hamming distances for NEBNext Illumina adaptors
Rights and permissions
About this article

Cite this article
Grzybowski, A.T., Shah, R.N., Richter, W.F. et al. Native internally calibrated chromatin immunoprecipitation for quantitative studies of histone post-translational modifications. Nat Protoc 14, 3275–3302 (2019). https://doi.org/10.1038/s41596-019-0218-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-019-0218-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
