
Abstract
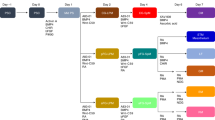
The human stomach contains two primary domains: the corpus, which contains the fundic epithelium, and the antrum. Each of these domains has distinct cell types and functions, and therefore each presents with unique disease pathologies. Here, we detail two protocols to differentiate human pluripotent stem cells (hPSCs) into human gastric organoids (hGOs) that recapitulate both domains. Both protocols begin with the differentiation of hPSCs into definitive endoderm (DE) using activin A, followed by the generation of free-floating 3D posterior foregut spheroids using FGF4, Wnt pathway agonist CHIR99021 (CHIR), BMP pathway antagonist Noggin, and retinoic acid. Embedding spheroids in Matrigel and continuing 3D growth in epidermal growth factor (EGF)-containing medium for 4 weeks results in antral hGOs (hAGOs). To obtain fundic hGOs (hFGOs), spheroids are additionally treated with CHIR and FGF10. Induced differentiation of acid-secreting parietal cells in hFGOs requires temporal treatment of BMP4 and the MEK inhibitor PD0325901 for 48 h on protocol day 30. In total, it takes ~34 d to generate hGOs from hPSCs. To date, this is the only approach that generates functional human differentiated gastric cells de novo from hPSCs.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
McCracken, K. W. et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014).
McCracken, K. W. et al. Wnt/beta-catenin promotes gastric fundus specification in mice and humans. Nature 541, 182–187 (2017).
Bartfeld, S. et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136.e6 (2015).
Bertaux-Skeirik, N. et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 11, e1004663 (2015).
Dedhia, P. H., Bertaux-Skeirik, N., Zavros, Y. & Spence, J. R. Organoid models of human gastrointestinal development and disease. Gastroenterology 150, 1098–1112 (2016).
Schlaermann, P. et al. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut 65, 202–213 (2016).
Willet, S. G. & Mills, J. C. Stomach organ and cell lineage differentiation: from embryogenesis to adult homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2, 546–559 (2016).
Zorn, A. M. & Wells, J. M. Molecular basis of vertebrate endoderm development. Int. Rev. Cytol. 259, 49–111 (2007).
Munera, J. O. et al. Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell 21, 51–64.e6 (2017).
Rankin, S. A. et al. Timing is everything: reiterative Wnt, BMP and RA signaling regulate developmental competence during endoderm organogenesis. Dev. Biol. 434, 121–132 (2018).
Stevens, M. L. et al. Genomic integration of Wnt/beta-catenin and BMP/Smad1 signaling coordinates foregut and hindgut transcriptional programs. Development 144, 1283–1295 (2017).
Roberts, D. J. et al. Sonic hedgehog is an endodermal signal inducing Bmp-4 and Hox genes during induction and regionalization of the chick hindgut. Development 121, 3163–3174 (1995).
Bayha, E., Jorgensen, M. C., Serup, P. & Grapin-Botton, A. Retinoic acid signaling organizes endodermal organ specification along the entire antero-posterior axis. PLoS ONE 4, e5845 (2009).
Molotkov, A., Molotkova, N. & Duester, G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev. Dyn. 232, 950–957 (2005).
Wang, Z., Dolle, P., Cardoso, W. V. & Niederreither, K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 297, 433–445 (2006).
Engevik, K. A., Matthis, A. L., Montrose, M. H. & Aihara, E. Organoids as a model to study infectious disease. Methods Mol. Biol. 1734, 71–81 (2018).
Takebe, T., Wells, J. M., Helmrath, M. A. & Zorn, A. M. Organoid center strategies for accelerating clinical translation. Cell Stem Cell 22, 806–809 (2018).
Finkbeiner, S. R. et al. Transcriptome-wide analysis reveals hallmarks of human intestine development and maturation in vitro and in vivo. Stem Cell Rep. 4, 1140–1155 (2015).
Kroon, E. et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 26, 443–452 (2008).
Si-Tayeb, K. et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51, 297–305 (2010).
Schumacher, M. A. et al. Helicobacter pylori-induced sonic hedgehog expression is regulated by NFκB pathway activation: the use of a novel in vitro model to study epithelial response to infection. Helicobacter 20, 19–28 (2015).
Wroblewski, L. E. et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 64, 720–730 (2015).
McCracken, K. W., Howell, J. C., Wells, J. M. & Spence, J. R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 6, 1920–1928 (2011).
Spence, J. R. et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011).
Workman, M. J. et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 23, 49–59 (2017).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Teo, A. K. et al. Activin and BMP4 synergistically promote formation of definitive endoderm in human embryonic stem cells. Stem Cells 30, 631–642 (2012).
Acknowledgements
We thank D. Kechele for comments on the manuscript. This work was supported by grants from the National Institutes of Health (R01DK092456, U19AI116491, and P01HD093363 to J.M.W.). We also acknowledge core support from the Pluripotent Stem Cell Facility of Cincinnati Children’s Hospital Medical Center. We acknowledge core support from a Cincinnati Digestive Disease Center award (P30DK0789392).
Author information
Authors and Affiliations
Contributions
J.M.W., K.W.M., and T.R.B. conceived the study and experimental design. T.R.B. and J.M.W. co-wrote the manuscript. T.R.B. produced all images for the figures, and J.M.W. produced the protocol schematic. K.W.M. and T.R.B. analyzed the data and performed experiments.
Corresponding author
Ethics declarations
Competing interests
J.M.W. and K.W.M. are listed on the following patent applications: PCT/US2015/032626, ‘Methods and systems for converting precursor cells into gastric tissues through directed differentiation’, and PCT/US2017/031309, ‘Methods for the in vitro manufacture of gastric fundus tissue and compositions related to same’. T.R.B. declares no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Key references using this protocol
McCracken, K. W. et al. Nature 516, 400–404 (2014): https://doi.org/10.1038/nature13863
McCracken, K. W. et al. Nature 541, 182–187 (2017): https://doi.org/10.1038/nature21021
Rights and permissions
About this article

Cite this article
Broda, T.R., McCracken, K.W. & Wells, J.M. Generation of human antral and fundic gastric organoids from pluripotent stem cells. Nat Protoc 14, 28–50 (2019). https://doi.org/10.1038/s41596-018-0080-z
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-018-0080-z
This article is cited by
-
Collagen-based biomaterials in organoid technology for reproductive medicine: composition, characteristics, and applications
Collagen and Leather (2023)
-
Mechanically enhanced biogenesis of gut spheroids with instability-driven morphomechanics
Nature Communications (2023)
-
Functional precision oncology using patient-derived assays: bridging genotype and phenotype
Nature Reviews Clinical Oncology (2023)
-
Directed differentiation of ureteric bud and collecting duct organoids from human pluripotent stem cells
Nature Protocols (2023)
-
3D and organoid culture in research: physiology, hereditary genetic diseases and cancer
Cell & Bioscience (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
