
Abstract
SARS-CoV-2 ORF3a is a putative viral ion channel implicated in autophagy inhibition, inflammasome activation and apoptosis. 3a protein and anti-3a antibodies are found in infected patient tissues and plasma. Deletion of 3a in SARS-CoV-1 reduces viral titer and morbidity in mice, suggesting it could be an effective target for vaccines or therapeutics. Here, we present structures of SARS-CoV-2 3a determined by cryo-EM to 2.1-Å resolution. 3a adopts a new fold with a polar cavity that opens to the cytosol and membrane through separate water- and lipid-filled openings. Hydrophilic grooves along outer helices could form ion-conduction paths. Using electrophysiology and fluorescent ion imaging of 3a-reconstituted liposomes, we observe Ca2+-permeable, nonselective cation channel activity, identify mutations that alter ion permeability and discover polycationic inhibitors of 3a activity. 3a-like proteins are found across coronavirus lineages that infect bats and humans, suggesting that 3a-targeted approaches could treat COVID-19 and other coronavirus diseases.
Similar content being viewed by others

Main
Coronavirus disease 2019 (COVID-19), caused by the SARS-CoV-2 virus, is an ongoing global pandemic. Vaccine and therapeutic development are predominantly focused on the essential virus-encoded Spike, main protease and RNA-dependent RNA polymerase proteins. High-resolution structures of these targets, some in complex with drug candidates or neutralizing antibodies, have yielded mechanistic insight into their function and have provided a platform for successful structure-guided vaccine and drug design1,2,3,4,5,6. Still, expanding our knowledge of SARS-CoV-2 target proteins remains important both for understanding SARS-CoV-2 virology and developing alternative treatments to mitigate against potential resistance that evolves, or new viruses that emerge, in the future7.
Virally encoded open reading frame 3a (ORF3a, 3a for short) has been considered a potential target for vaccines or therapeutics8,9. 3a is highly conserved within the Betacoronavirus subgenus Sarbecovirus, which includes SARS-CoV-1 and other related bat coronaviruses (Supplementary Fig. 1)10. Biopsies from SARS-CoV-1 patients show 3a expression in infected tissue and SARS-CoV-1 and SARS-CoV-2 patient plasma contain anti-3a antibodies8,11,12,13. SARS-CoV-1 3a has been implicated in inflammasome activation14 and both apoptotic15 and necrotic cell death16 while SARS-CoV-2 3a has been implicated in apoptosis and inhibition of autophagy in vitro17. In mouse models of both SARS-CoV-1 and 2 infection, genomic deletion of 3a reduces viral titer and morbidity18,19.
3a has been proposed to form an ion channel and is one of two putative viroporins encoded by the SARS-CoV-2 genome together with the envelope protein E (SARS-CoV-1 encodes an additional putative viroporin, ORF8a). SARS-CoV-1 3a has been reported to form an emodin- and Ba2+-sensitive cation channel20,21. Viroporins from other viruses have been shown to modify host membrane permeability to promote viral assembly and release22,23,24. However, the precise roles of 3a in disease pathogenesis are unknown, precluded in part by the lack of a mechanistic understanding of 3a. Here, we determine structures of SARS-CoV-2 3a by cryo-electron microscopy (cryo-EM) and assess putative ion channel activity in vitro using electrophysiology and fluorescent ion flux assays.
Results
Structure determination
We determined structures of dimeric and tetrameric SARS-CoV-2 3a in lipid nanodiscs. Full length SARS-CoV-2 3a was heterologously expressed in Spodoptera frugiperda (Sf9) cells with a cleavable C-terminal green fluorescent protein (GFP) tag. Purification of 3a in detergent resulted in two species separable by gel filtration (Extended Data Fig. 1). Most 3a runs at a position consistent with a 62-kDa dimer (Extended Data Fig. 1a,c,e) and roughly 10–30% runs as a 124-kDa tetramer (Extended Data Fig. 1). A similar degree of tetramer formation was observed at low concentrations of 3a by fluorescence size-exclusion chromatography, indicative of a biochemically stable species rather than concentration-dependent nonspecific aggregation (Extended Data Fig. 1e). These data are consistent with previous reports of dimeric and tetrameric SARS-CoV-1 3a observed by western blot16,20.
We separately reconstituted dimeric and tetrameric SARS-CoV-2 3a into nanodiscs made from the scaffold protein MSP1E3D1 and a mixture of DOPE, POPC and POPS lipids, and determined their structures by cryo-EM to 2.9- and 6.5-Å resolution (Extended Data Figs. 2–4, Supplementary Fig. 2 and Table 1). Based on a report of improved resolution of apoferritin and GABAA structures using new cryo-EM instrumentation (including the electron source, energy filter and camera; see Methods for details)25, we asked whether similar improvements would be possible for the sub-100-kDa 3a. Indeed, a reconstruction generated from data collected on this instrumentation, using the same batch of grids, was improved to 2.1 Å (Fig. 1, Extended Data Figs. 2 and 5, Supplementary Fig. 4 and Table 1). We note we cannot exclude other differences including in accelerating voltage, pixel size or defocus range from potentially contributing to the improvement in resolution.

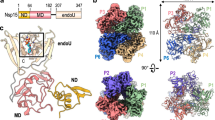
a, Cryo-EM map of the 3a dimer in MSP1E3D1 nanodiscs at 2.1-Å nominal resolution, viewed from the membrane plane. One subunit is colored gray, and the second subunit is colored with transmembrane region in orange and the CD in pink. Density from the nanodisc MSP1E3D1 is colored tan, and the DOPE lipid is coral. b,c, Model of dimeric 3a viewed from the membrane (b) (as in a) and from the extracellular or lumenal side (c). d, Enlarged view of the interaction between subunits in the CD with residues forming the hydrophobic core indicated. e, Schematic of a 3a monomer. Secondary structure elements are indicated, and unmodeled termini and a five-amino-acid β3–β4 loop are shown with dashed lines. f, Cryo-EM density and model for a selected strand from the CD at two different thresholds. g, Enlarged view of density in the MSP1E3D1-interaction region. h, Model in the same region as g with key residues in the area displayed as sticks.
3a dimer structure
The 2.9-Å dimeric reconstruction (with C2 symmetry applied, PDB 6XDC) permitted de novo modeling of 195 of the 275 amino acids per protomer chain. The N terminus (amino acids 1–39), C terminus (amino acids 239–275) and a short cytoplasmic loop (amino acids 175–180) are weakly resolved in the density map due to conformational differences between particles or because they are disordered (Supplementary Fig. 2 and Extended Data Fig. 3). The 2.1-Å dimeric reconstruction (with C2 symmetry applied, PDB 7KJR) allowed for improved rotamer assignments due to better defined side chain density throughout the protein, including visible holes in many aromatic residues (Fig. 1a–f). We additionally modeled a portion of each MSP1E3D1 scaffold protein (amino acids 25–55, Fig. 1g,h), two DOPE lipids and 122 water molecules. We refer to the higher-resolution structure in the remainder of the paper unless otherwise indicated. The scaffold protein is partially well-resolved due to a specific interaction visible on each side of the 3a dimer (even in C1 reconstructions) between MSP1E3D1 W40 and the transmembrane region of 3a (Fig. 1g,h). The symmetric nature of this interaction means that the two MSPE3D1 protomers must twist around the lipid bilayer rather than adopting the canonical arrangement of two parallel stacked rings.
3a adopts a new fold that, to our knowledge, has not been observed in available protein structures. Querying the protein structure database for homologs with Dali returned only weak hits for fragments of 3a domains26. Viewed from the membrane plane, 3a is approximately 70 Å tall with a 40-Å high transmembrane region and a cytosolic domain (CD) extending 30 Å out of the membrane (Fig. 1a,b). The transmembrane region is composed of three helices per protomer with N termini oriented on the extracellular or lumenal side and C termini on the cytosolic side of the membrane. Viewed from the extracellular side, the transmembrane helices (TMs) trace the circumference of an ellipse with TM1–3 from one protomer followed by TM1–3 of the second protomer in a clockwise order (Fig. 1c). TM1s and TM2s pack against each other across the elliptical minor axis with TM3s positioned at the main axis vertices. TM1–TM2 and TM2–TM3 are joined by short intracellular and extracellular linkers, respectively.
The transmembrane region connects to the CD through a turn-helix-turn motif following TM3. Each protomer chain forms a pair of opposing β-sheets packed against one another in an eight stranded β-sandwich (Fig. 1b,d). The outer sheet is formed by strands β1, β2 and β6 and the N-terminal half of β7. The inner sheet is formed by strands β3, β4, β5 and β8 and the C-terminal half of β7. The inner sheets from each protomer interact through a large (roughly 940 Å2 of buried surface area per chain) and highly complementary interface with residues V168, V225, F230 and I232 forming a continuous hydrophobic core (Fig. 1d). The interaction between β-sandwiches from each protomer thus forms a strong and stable link between monomers in the dimer.
Structural features of 3a tetramers
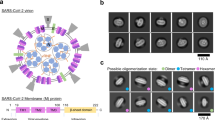
We next examined the structure of 3a tetramers. Two-dimensional (2D) class averages of tetrameric 3a show a side-by-side arrangement of two dimers with separated TMs and close juxtaposition of CDs (Fig. 2a). Tetrameric 3a reconstructions had lower final resolutions (roughly 6.5 Å) than dimeric 3a (Fig. 2a and Extended Data Fig. 2), which precluded detailed model building, but were sufficiently featured in the CDs for rigid-body docking of two copies of the 3a dimer to determine their approximate relative orientation (Fig. 2b and Extended Data Fig. 6a). The best fit model shows a continuous interface (roughly 300 Å2 buried surface area per dimer) formed between TM3–CTD linkers and β1–β2 linkers from neighboring dimers. While the exact positions of side chains cannot be determined at this resolution, residues W131, R134, K136, H150, T151, N152, C153 and D155 are poised to mediate tetramerization through a network of hydrophobic, polar and electrostatic interactions (Fig. 2c). In SARS-CoV-1 3a, reducing agents and a C133A mutation resulted in loss of oligomerization, membrane localization and ion channel activity20. However, expression of the C133A mutant was dramatically reduced, suggesting these results might be a consequence of protein destabilization20. In SARS-CoV-2 3a, C133 is located in a notable cysteine-rich pocket adjacent to the tetramerization interface (Extended Data Fig. 6b). At the base of TM3, C133 projects back toward the top faces of β1 and β2 in close proximity to solvent exposed C148 and buried C157. All three cysteines are reduced in the structure, although they are within potential disulfide-bonding distance (Cα distance 4.4–6.5 Å) (Extended Data Fig. 6b). While it is unlikely a disulfide involving C133 mediates tetramerization of 3a without substantial rearrangement of this region, it could be that disruption of this cysteine-rich pocket with cysteine modifying agents or mutations disfavors 3a oligomerization allosterically.

a, Two-dimensional class average of tetrameric 3a in MSP1E3D1 lipid nanodiscs (top) and cryo-EM map at 6.5-Å nominal resolution (bottom). b, Approximate assembly of 3a tetramers. Two 3a dimer structures are rigid-body fit into the tetrameric cryo-EM map. c, Enlarged view of the interface between 3a dimers observed after rigid-body fitting with residues potentially mediating the interaction displayed as sticks.
Potential ion permeation paths through 3a
Ion conduction pathways are required for ion channel function, so we analyzed the 3a structures to identify potential permeation routes. 3a forms a large polar cavity within the inner half of the TM region. The cavity is continuous with the cytosol and surrounding bilayer through three pairs of openings: the upper, lower and intersubunit tunnels (Fig. 3a–f). The upper tunnels are formed between TM2 and TM3 within each protomer, narrow to roughly 2.2 Å in radius, and likely open to the membrane (Fig. 3a–c). The lower tunnels run underneath the TM1–TM2 linker and above the CD, narrow to roughly 2 Å in radius, and open to the cytosol (Fig. 3a,d,e). The lower tunnels are open paths for ion and water movement between the cell interior and channel cavity. Consistently, a network of ordered water molecules extends through these openings, the cavity and the CD (Fig. 3b,d). The intersubunit tunnels run between TM1 and TM3 from opposing protomers, just above the CD, and open to the membrane–cytosol interface (Fig. 3a,f). Strong lipid-shaped density present within each intersubunit tunnel is modeled as DOPE (the most abundant lipid in these nanodiscs) with the ethanolamine head group pointed into the cavity. Lipid binding is stabilized by electrostatic interactions: the positively charged ethanolamine interacts with negatively charged D142 and the backbone carbonyls of I63 and D142 while the lipid phosphate interacts with R126, R122, Y206, N144 and the backbone amide of L65.

a, View of a 3a dimer from the membrane plane with key regions labeled and with planes (dotted lines) and viewing direction (arrow) for g–i) indicated. b, The 3a upper tunnel with solvent-excluded surface shown for each subunit and water molecules shown as light blue spheres. The approximate lipid bilayer region is marked by black lines. One 3a subunit is colored orange and the other in gray. c, Left, model of 3a (gray) with the HOLE path through the upper tunnel shown in transparent cyan. Lipid (coral) is displayed as sticks. Middle, the radius of the upper tunnel as a function of distance from the cavity center calculated with HOLE. Right, the view into the upper tunnel from the membrane with key residues shown as sticks. d, As in b, for a view of the lower tunnel. e, As in c, for the lower tunnel, with the view on the right into the tunnel from the cytosol. f, The intersubunit tunnel with bound DOPE lipid (coral) shown as space-filling spheres. Top right, an enlarged view of the lipid interaction with key residues shown as sticks. Bottom-right, a cut-through view of the cryo-EM density of the lipid interaction region viewed from the cytosolic side. g–i, Model and transparent surface of the viewing planes indicated in a (g, view G; h, view H; i, view I). Water molecules are shown as light blue spheres. One 3a subunit is colored orange and the other in gray. j, The solvent-excluded surface of 3a colored from hydrophilic (dark cyan) to most hydrophobic (dark orange). A membrane-facing hydrophilic groove is outlined. k, Key residues in the hydrophilic groove are depicted as sticks colored in dark cyan.
While the polar cavity reduces the energetic barrier to ion movement across the inner half of the low dielectric membrane, the current structures lack a central pore connecting the cavity to the lumenal or extracellular side. Instead, a hydrophobic seal is formed between TMs in the extracellular half of the membrane above the polar cavity (Fig. 3). The top of the cavity is formed by polar interactions between Q57, S58, N82 and Q116 side chains (Fig. 3a,g). Just extracellular to these interactions, the hydrophobic seal is formed by interactions between residues F43, L46, I47, V50, L95 and L96 (Fig. 3a,h,i). Opening a central pore would require substantial conformational rearrangement to break these hydrophobic interactions.
Most ion channels have central pores for conduction, but some, including those of the TMEM16 and OSCA families, have evolved ion pathways through external grooves or tunnels near membrane-facing surfaces27,28,29,30. 3a exhibits a distinctive membrane-facing, partially hydrophilic groove is observed between TM2 and TM3 (Fig. 3j). It is bounded by the upper tunnel and TM2–TM3 linker at the cytoplasmic and extracellular sides, respectively, and is lined by conserved polar residues N119, N82, S117, Q116, S58, T89, Y113, S92, Y109 and H93 (Fig. 3k). The hydrophilic character of this region would be expected to lower the energetic barrier for movement of ions across the outer half of the membrane and more subtle conformational changes would be required to permit permeation along this route relative to a central pore. 3a oligomerization could also be important for building a conduction path either through conformational changes or pore assembly, as was proposed for SARS-CoV-1 3a (ref. 20). While a pore is not formed in the context of the SARS-CoV-2 tetramer, we observe that one of the partially hydrophilic TM2–TM3 grooves from each dimeric unit face one another across a narrow membrane (Extended Data Fig. 6c).
Assessment of channel activity in 3a-reconstituted proteoliposomes
We next used electrophysiology and calcium imaging to assess whether SARS-Cov-2 3a is capable of permeating ions. We did not observe consistent currents in recordings from transfected cells and reasoned this could be due to most of 3a being localized to internal membranes (Extended Data Fig. 7), inactivation at the plasma membrane and/or 3a toxicity. We therefore purified 3a in detergent, reconstituted it into phosphatidylcholine-containing proteoliposomes, and recorded currents across excised patches pulled from proteoliposome blisters. Excised patches from 3a-containing liposomes exhibited currents that reversed at 0 mV and displayed modest outward rectification in symmetric [K+] (Fig. 4a and Extended Data Fig. 8). In contrast, channel-like activity was not observed in recordings from mock-reconstituted (empty) proteoliposomes (Extended Data Fig. 8g). We evaluated ion selectivity of 3a by replacing the K+-containing bath solution with solutions containing Na+, NMDG+ or Ca2+. Solution exchange resulted in reversal potential shifts from 0.3 ± 0.3 mV in K+ to −8.2 ± 0.8 mV in Ca2+, −13.5 ± 1.8 mV in Na+ and −31.0 ± 1.1 mV in NMDG+ (Fig. 4b,d). These shifts correspond to the following permeability ratios (PX/PK+): Ca2+ (1.88 ± 0.08) > K+ (1.0 ± 0.01) > Na+ (0.59 ± 0.04) > NMDG+ (0.29 ± 0.01) (Fig. 4c). Alkaline or acidic pH had little effect on channel activity (Extended Data Fig. 8f). Consistent with the electrophysiological measurements, we also observed significant calcium influx in 3a proteoliposomes using Fluo-5N calcium imaging following the addition of 8 mM [Ca2+]ext compared to empty liposomes (Fig. 4e).

a, Current–voltage relationship from a 3a-proteoliposome patch. Pipette solution was 150 mM K+ and external solution was 150 mM K+ (black), 150 mM Na+ (pink), 75 mM Ca2+ (orange) or 150 mM NMDG+ (blue) (mean ± s.e.m., n = 3 recordings from a representative patch). b, Reversal potential. Pipette solution was 150 mM K+ and external solution was 150 mM K+ (black), 150 mM Na+ (pink), 75 mM Ca2+ (orange) or 150 mM NMDG+ (blue) (mean ± s.e.m., n = 4, 4, 12 and 4 patches, respectively). One-way ANOVA with Dunnett correction, ****P < 0.0001). c, Permeability ratios (PX+/PK+) calculated from b. Pipette solution was 150 mM K+ and external solution was 150 mM K+ (black), 150 mM Na+ (pink), 75 mM Ca2+ (orange) or 150 mM NMDG+ (blue) (mean ± s.e.m., n = 4, 4, 12 and 4 patches, respectively). One-way ANOVA with Dunnett correction, *P < 0.05, ***P < 0.001, ****P < 0.0001). d, Gap-free current recording held at −80 mV during bath solution exchanges indicated in the bar above the current trace. e, Histogram of peak calcium influx (Fmax/F) measured by Fluo-5N fluorescence following the addition of 8 mM [Ca2+]ext to 3a liposomes (blue) or empty liposomes (gray). For the inset graph, mean ± s.e.m. of peak calcium influx for 3a liposomes, empty liposomes and 3a liposomes incubated in 200 μM ruthenium red (one-way ANOVA (F(2,4945) = 99.01); n = 1,219, 2,178 and 1,551 liposomes per group, respectively, with Dunnett’s multiple-comparisons test, ****P < 0.0001). f, Gap-free current recording in symmetric 150 mM KCl held at −80 mV during bath solution exchanges of varying ruthenium red concentrations indicated in the bar below the current trace. The boxed inset shows magnified channel openings and closures selected from the region indicated. g, Normalized activity in symmetric 150 mM KCl at different concentrations of ruthenium red with fit (black line) and 95% confidence interval (red) shown (mean ± s.e.m., n = 3 patches, IC50 = 90 ± 10 μM). h, Normalized 3a activity in symmetric 150 mM KCl and (from left to right) no blocker, 1 μM ruthenium red (P = 0.9127), 10 μM ruthenium red (P < 0.001), 100 μM ruthenium red (P < 0.0001), 500 μM ruthenium red (P < 0.0001), 1 mM spermidine (P < 0.0001),, 10 mM spermidine (P < 0.0001), 0.1 mM Gd3+ (P = 0.0441), 1 mM Gd3+ (P < 0.0001), 10 mM Gd3+ (P < 0.0001), 1 mM Fe3+ (P < 0.0001) and 1 mM Tb3+ (P < 0.0001) (mean ± s.e.m., n = 3 patches for each, one-way ANOVA with Dunnett correction, *P < 0.05, ***P < 0.001, ****P < 0.0001). NS, not significant. Source data for a–c,e,g and h are available online.
We performed mass spectrometry of 3a samples to assess whether channel activity observed in vitro could be due to channel contaminants in purified 3a or lipids used for reconstitution. We analyzed samples of 3a in detergent before liposome incorporation, 3a-reconstituted proteoliposomes and mock-reconstituted liposomes, and searched a database consisting of the 3a construct, the S. frugiperda (expression system) proteome, the Glycine max (lipid source) proteome and common contaminants (Extended Data Fig. 9). We detected 3a peptides in both 3a-containing samples, with the detergent sample giving better sequence coverage and more matched peptides. The only other proteins identified were at low levels and from common contaminants; no peptides from known channels or transmembrane proteins other than 3a were observed in any sample. While we cannot definitively rule out the presence of an unannotated contaminant or one below the detection threshold for mass spectrometry, these results indicate that SARS-CoV-2 3a is responsible for the Ca2+-permeable nonselective channel activity observed in vitro.
Polycations block 3a channel activity in vitro
We next asked whether blockers of nonselective cation channels inhibit 3a ion conduction in liposomes. We found that ruthenium red, a 786-Da polycationic dye, blocks 3a activity in current recordings (Fig. 4g,h) and Ca2+ influx measurements (Fig. 4e). Ruthenium red displays dose-dependent inhibition of 3a activity in proteoliposome recordings (IC50 = 90 ± 10 µM) with flickery block at negative potentials, similar to that observed with other large pore channels (Fig. 4g). In addition, we identified the polyamine spermidine, which showed near complete block at 10 mM, and the trivalent ions Gd3+, Fe3+ and Tb3+, which showed partial block at 1–10 mM, as 3a inhibitors in proteoliposome recordings (Fig. 4g). ruthenium red, spermidine, Gd3+ and other trivalents are relatively nonselective inhibitors of cation channels including TRPs, RyRs, CALHMs, K2Ps and KIRs31,32,33,34,35,36,37, but the spectrum of 3a blockers and affinities observed here is, to our knowledge, distinct from those of other known channels. We further tested reported low-affinity blockers of SARS-CoV-1 3a channel activity20,21, but found that neither Ba2+ nor the small molecule emodin inhibited SARS-CoV-2 3a activity (Extended Data Fig. 8d,e). Consistently, in a cryo-EM structure of dimeric 3a determined in the presence of 100 µM emodin to 3.7-Å resolution (Extended Data Figs. 2 and 4, Supplementary Fig. 3 and Table 1), we observed no evidence of emodin binding or structural changes compared to apo 3a.
3a permeates the large divalent ion YO-PRO-1
Given the ability of 3a channels to weakly conduct NMDG+ (Fig. 4a–d), we next used a YO-PRO-1 fluorescence-based flux assay to assess the ability of 3a channels to conduct other large cations. YO-PRO-1, a 629-Da divalent cation, does not readily cross lipid bilayers and has been used to study the activity of other nonselective cation channels, including P2X7 and TRP channels, that can conduct large organic cations31,32,38,39. Robust YO-PRO-1 uptake was observed in a subset of 3a liposomes that was significantly higher than background fluorescence observed in empty liposomes (Fig. 5a–d). To compare YO-PRO-1 uptake across multiple experiments, we quantified a single value (area under the curve) to represent both the number of YO-PRO-1+ liposomes and the amount of YO-PRO-1 uptake (Fig. 5e). Using this analysis, we found 3a liposomes display significantly greater YO-PRO-1 uptake and accumulation as compared to empty liposomes (Fig. 5e). Uptake was dependent on 3a protein because liposomes containing the human K2P channel TRAAK displayed little uptake and accumulation of YO-PRO-1, similar to empty liposomes, as expected for a highly selective K+ channel (Fig. 5a,c–e). Similar to current recordings (Fig. 4e,f), we observed a dose-dependent ruthenium red block of 3a activity in the YO-PRO-1 flux assay (IC50 = 175 ± 98 µM). These data indicate that 3a can form an ion channel that is capable of passing large cations. Ruthenium red (786 Da) block and YO-PRO-1 (629 Da) and NMDG+ (406 Da) permeation sets limits on 3a pore size that are similar those observed for TRP and P2X channels.

a–c, Histograms from quantified images following a 10-min incubation in 10 μM YO-PRO-1 for empty liposomes (a), 3a liposomes (b) or TRAAK liposomes (c) (n = 255, 3,046 and 1,408 for empty, 3a and TRAAK liposomes, respectively). Dotted red lines indicate the position of the mean fluorescence of empty liposomes. Insets, representative images with YO-PRO-1 fluorescence (green) overlaid on brightfield. Scale bars, 20 μm. d, Fraction of YO-PRO-1-positive liposomes (mean ± s.d., n = 9, 10 and 10 independent replicates for empty, 3a and TRAAK liposomes with 500–5,000 liposomes per replicate, P = 0.0004 (one-way ANOVA (F(2,26) = 10.52)). Tukey’s multiple-comparisons test Pempty versus 3a = 0.0004; PTRAAK versus 3a = 0.0113 and Pempty versus TRAAK = 0.3571). e, Area under the curve (AUC) of YO-PRO-1 fluorescence histograms of the fraction of liposomes that took up YO-PRO-1 per well (mean ± s.d., n = 9, 10 and 10 independent replicates for empty, 3a and TRAAK liposomes with 500–5,000 liposomes per replicate, P < 0.0001 (Welch’s ANOVA test (W(2,16.68) = 13.22)), Dunnett’s multiple-comparisons test: Pempty versus 3a = 0.0005; PTRAAK versus 3a = 0.0041; Pempty versus TRAAK = 0.4178). f, YO-PRO-1 uptake in 3a liposomes is inhibited by ruthenium red. Mean ± s.d., sigmoidal fit (black dotted line) and IC50 are shown. Source data for a–f are available online.
3a mutants alter ion conduction
We speculate that, relative to the structures presented here, an open conformation of 3a involves substantial rearrangement of the hydrophobic seal to create a central pore or more subtle rearrangement of the partially hydrophilic membrane-facing grooves between TM2 and TM3 to create lateral conduction pathways. To probe the structural determinants for 3a ion conduction, we set out to identify 3a mutations that alter channel activity. To this end, we purified nine mutated or truncated 3a constructs, reconstituted each into proteoliposomes, and compared them to wild-type 3a in electrophysiological recordings (Fig. 6a and Extended Data Fig. 10). We find that two separate mutants alter channel activity, one at the top of the cavity (Q57E) and another and base of the TM2–TM3 grooves (S58L,Q116L). Both Q57E and S58L,Q116L mutants reduce Ca2+ and NMDG+ permeability without altering Na+ or K+ permeability. These effects were specific to these mutations as seven additional mutations, including a common circulating variant Q57H40, had no effect on relative ion permeability. These results are consistent with the hypothesis that 3a channel opening involves conformational changes of residues that form constrictions above the polar cavity or along the hydrophilic membrane-facing TM2–TM3 grooves.

a, Two views of 3a from the membrane plane with the positions of 3a mutations and truncation indicated. b, Permeability ratios (PX/PK+) calculated from reversal potential shifts for 3a mutations and ΔN truncation (mean ± s.e.m., n = 8, 3, 3, 7, 2, 4, 7, 8, 9 and 3 patches for wild-type (WT), S58K (P = 0.2352), C81E (P = 0.1741), N119K (P = 0.9971), D142K (P = 0.9997), Q57H (P = 0.9005), Q57K (P = 0.2220), Q57E (P < 0.0001), S58L,Q116L (P < 0.0001) and ΔN (for Ca2+ P = 0.0251 and for NMDG+ P = 0.0287), respectively, *P < 0.05, ****P < 0.0001, one-way ANOVA with Dunnett correction for multiple comparisons). Source data for b are available online.
Our structural analysis revealed an unassigned region of density in the cryo-EM maps stretching between subunits just above the extracellular side of the transmembrane region and near the top of the TM2–TM3 grooves that likely corresponds to a portion of the unmodeled N terminus (Extended Data Fig. 7). To investigate the potential role of the N terminus in channel function, we generated an N-terminal deletion construct lacking the first 41 amino acids (3aΔN). This mutant displayed reduced Ca2+ and NMDG+ relative permeability in proteoliposome recordings, albeit to a lesser extent than Q57E and S58L,Q116L (Fig. 6b). The relative permeability changes observed in liposomes suggest that residues in the N terminus, such as those at the top of the polar cavity, mediate the ability of 3a to permeate large cations. In addition, the N terminus may be involved in 3a subcellular localization as 3aΔN channels displayed increased localization to the plasma membrane (Extended Data Fig. 7).
3a-like proteins in Alpha- and Betacoronavirus
While 3a is well conserved in the Betacoronavirus subgenus (Supplementary Fig. 1), related proteins have not yet been identified in other coronaviruses, including the other five species known to infect humans: MERS-CoV, HCoV-NL63, HCoV-229E, HCoV-HKU1 and HCoV-OC43. Thus, we asked whether we could identify more distant homologs using the 3a structure and structure prediction algorithms. 3a homologs were not detected in Gammacoronavirus, Deltacoronavirus or in the Betacoronavirus subgenus Embecovirus (which includes HCoV-HKU1 and HCoV-OC43). Distant homology to the CD was identified in the membrane protein ORF5 found in Betacoronavirus subgenus Merbecovirus species including MERS-CoV. We identified high confidence structural homologs in all remaining Betacoronavirus and Alphacoronavirus subgenera (including in HCoV-229E and HCoV-NL63 (Supplementary Fig. 5)), several of which have also been reported to have ion channel activity41,42,43. Notably, coronaviruses with 3a homologs have been proposed to derive from viruses that circulate primarily in bats, while coronaviruses without 3a structural homologs have been proposed to derive from viruses that circulate primarily in rodents, birds or pigs (Table 2). It may be that the presence of 3a in coronaviruses with bats as their principal reservoir reflects a unique aspect of bat coronavirus biology.
Discussion
Together, our data demonstrate that SARS-CoV-2 3a adopts a novel dimeric fold widely conserved across Coronaviridae and can form a nonselective Ca2+ permeable cation channel. These results provide a basis for further studies to determine mechanisms of 3a channel gating and conduction. A polar cytoplasmic cavity creates a low energy path for ions to traverse between the cytoplasm and halfway across the membrane. However, 3a lacks a central pore across the outer half of the membrane and conformational changes to open one would require breaking interactions that form a hydrophobic seal between TMs in the current structure. Alternatively, lateral conduction pathways could be formed along conserved hydrophilic membrane-facing grooves between TM2 and TM3. Similar lateral conduction paths have precedence among other channels including members of the TMEM16 and OSCA families27,28,29,30.
Oligomeric 3a assemblies could also mediate 3a activation. 3a dimers associate into tetramers and assembly could feasibly be propagated further into higher-order oligomers. Consistently, we observe increased oligomeric 3a bands in SDS–PAGE gels after liposome incorporation (Extended Data Figs. 1 and 10). This observation, together with the lipid binding pocket within 3a and its localization to organellar membranes, suggests the membrane environment may be important for 3a structure and function.
In liposomes, SARS-CoV-2 3a exhibits permeability to large cations including NMDG+ and YO-PRO-1 and sensitivity to ruthenium red and Gd3+, broadly reminiscent of other Ca2+-permeable channels including TRPV1, TRPA1 and P2X7 (refs. 31,32) while displaying, to our knowledge, a unique functional fingerprint. We note that our data are only partially consistent with previously reported properties of SARS-CoV-1 3a. For example, we do not observe block by Ba2+ or emodin and while both proteins apparently form nonselective cation channels, their relative permeability for cations differs. This could be explained by sequence and functional divergence: 3a in the two viruses shares just 73% sequence identity. Alternatively, different recording and lipid environments might alter channel properties. SARS-CoV-1 3a activity was recorded in Xenopus oocytes or cultured cells, while SARS-CoV-2 3a activity was recorded in reconstituted membranes. Whether SARS-CoV-2 3a channel activity is observed in cells, perhaps in intracellular membranes where most heterologously expressed protein is localized, remains to be determined.
If 3a has channel activity in cells, it could be important for promoting viral maturation through inhibition of autophagy and disruption of lysosomes44,45. The unresolved N and C termini may also play important roles in determining 3a’s localization and binding partners. The calcium permeability of 3a is of particular interest in the context of infection. SARS-CoV-2 3a has been recently shown to trigger programmed cell death in cultured cells17 and calcium influx through 3a could serve as a switch that activates calcium-dependent caspases and apoptosis. Previous studies showed SARS-CoV-1 3a expression in infected lung pneumocytes, a key cell type also infected by SARS-CoV-2, and calcium signaling in type II pneumocytes plays an important role in maintaining airway homeostasis46,47. Thus, the expression of a calcium permeable channel, as for SARS-CoV-2 3a, could affect lung homeostasis and COVID-19 pathogenesis. Our data indicate that 3a could represent a target for treating COVID-19 and other coronavirus diseases and points the way to future experiments to elucidate the role of 3a in the viral life cycle and disease pathology.
Methods
Cloning and protein expression
The coding sequence for the 3a protein from SARS-Cov-2 was codon optimized for S. frugiperda (Sf9 cells) and synthesized (Integrated DNA Technologies). The sequence was then cloned into a custom vector based on the pACEBAC1 backbone (MultiBac, Geneva Biotech) with an added C-terminal PreScission protease (PPX) cleavage site, linker sequence, superfolder GFP (sfGFP) and 7×His tag, generating a construct for expression of 3a-SNS-LEVLFQGP-SRGGSGAAAGSGSGS-sfGFP-GSS-7×His. Mutants and truncation were also introduced into this construct using PCR. MultiBac cells were used to generate a Bacmid according to the manufacturer’s instructions. Sf9 cells were cultured in ESF 921 medium (Expression Systems) and P1 virus was generated from cells transfected with Escort IV reagent (MillaporeSigma) according to the manufacturer’s instructions. P2 virus was then generated by infecting cells at 2 million cells per ml with P1 virus at a multiplicity of infection of roughly 0.1, with infection monitored by fluorescence and harvested at 72 h. P3 virus was generated in a similar manner to expand the viral stock. The P2 or P3 viral stock was then used to infect Sf9 cells at 4 million cells per ml at a multiplicity of infection of around 2–5. At 72 h, infected cells containing expressed 3a-sfGFP protein were collected by centrifugation at 2,500g for 10 min and frozen at −80 °C.
Protein purification
For preparation of the 3a dimer and mutant constructs, infected Sf9 cells from 1 l of culture (roughly 15–20 ml of cell pellets) were thawed in 100 ml of lysis buffer containing 50 mM Tris, 150 mM KCl, 1 mM EDTA pH 8. Protease inhibitors (final concentrations E64 (1 µM), pepstatin A (1 µg ml−1), soy trypsin inhibitor (10 µg ml−1), benzamidine (1 mM), Aprotinin (1 µg ml−1), leupeptin (1 µg ml−1), AEBSF (1 mM) and phenylmethylsulfonyl fluoride (1 mM)) were added to the lysis buffer immediately before use. Benzonase (4 µl) was added after the cell pellet thawed. Cells were then lysed by sonication and centrifuged at 150,000g for 45 min. The supernatant was discarded and residual nucleic acid was removed from the top of the membrane pellet using Dulbecco’s phosphate buffered saline (DPBS). Membrane pellets were scooped into a dounce homogenizer containing extraction buffer (50 mM tris, 150 mM KCl, 1 mM EDTA, 1% n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace), pH 8). A 10% stock solution of DDM was dissolved and clarified by bath sonication in 200 mM tris pH 8 before addition to buffer to the indicated final concentration. Membrane pellets were then homogenized in extraction buffer and this mixture (150 ml of the final volume) was gently stirred at 4 °C for 1 h. The extraction mixture was centrifuged at 33,000g for 45 min and the supernatant, containing solubilized membrane protein, was bound to 4 ml of sepharose resin coupled to anti-GFP nanobody for 1 h at 4 °C. The resin was then collected in a column and washed with 10 ml of buffer 1 (20 mM HEPES, 150 mM KCl, 1 mM EDTA, 0.025% DDM, pH 7.4), 40 ml of buffer 2 (20 mM HEPES, 500 mM KCl, 1 mM EDTA, 0.025% DDM, pH 7.4) and 10 ml of buffer 1. The resin was then resuspended in 6 ml of Buffer 1 with 0.5 mg of PPX protease and rocked gently in the capped column for 2 h. Cleaved 3a protein was then eluted with an additional 8 ml of Wash Buffer, spin concentrated to roughly 500 µl with Amicon Ultra spin concentrator 10-kDa cutoff (Millipore), and then loaded onto a Superdex 200 increase column (GE Healthcare) on an NGC system (Bio-Rad) equilibrated in buffer 1. Peak fractions containing 3a channel were then collected and spin concentrated before incorporation into proteoliposomes or nanodiscs. For the tetramer, the preparation was carried out in a similar manner, except with overnight protease cleavage and collection of a peak of larger hydrodynamic radius (Extended Data Fig. 1). Traces for gel filtration were prepared using Chromlab 6.0 (Bio-Rad), Prism 8 and Adobe Illustrator.
Proteoliposome formation
For proteoliposome patching experiments, we incorporated protein into lipid and generated proteoliposome blisters for patch recordings using dehydration and rehydration as described previously with the following modifications48. The 3a dimer was first purified into buffer 1. Protein was then exchanged into lipid with the addition of Biobeads SM2 (Bio-Rad) and an overnight incubation at a protein:lipid ratio of 1:10 (corresponding to 0.5 mg purified 3a dimer and 5 mg of cleared soybean l-α-phosphatidylcholine, Soy PC, MillaporeSigma) in DR buffer (5 mM HEPES, 200 mM KCl, pH 7.2). For the YO-PRO-1 assay, 3a was incorporated at a ratio of 1:50. TRAAK control proteoliposomes were prepared at 1:50 as described previously48. Control liposomes were prepared from the same lipid mix and protocol with protein replaced with buffer 1.
Electrophysiology
All electrophysiology recordings were made from 3a-reconstituted Soy PC proteoliposomes. Patches formed in an inside-out configuration and were quickly (within 5–10 s) transferred to a solution exchange chamber. Recordings were made at room temperature using Clampex v.10.7 data acquisition software (as part of the pClamp v.10.7 suite) with an Axopatch 200B Patch Clamp amplifier and Digidata 1550B digitizer (Molecular Devices) at a bandwidth of 1 kHz and digitized at 500 kHz. A pressure clamp (ALA Scientific) was used to form seals. Potassium pipette and bath solution was 5 mM HEPES pH 7.2 and 150 mM KCl. Sodium bath solution was 5 mM HEPES pH 7.2 and 150 mM NaCl. NaCl in the bath solution was substituted for 150 mM NMDG-Cl or 75 mM CaCl2 for permeability experiments. Borosilicate glass pipettes were pulled and polished to a resistance of 2–5 MΩ when filled with pipette solution. For cation permeability experiments, liquid junction potentials were calculated and data were corrected offline. For current–voltage plots, the following voltage protocol was applied: Vhold = 0 mV mV; Vtest = −100 to +100 mV, Δ20 mV, ttest = 1 s. Currents from each patch correspond to mean values during the step to the indicated voltage.
Permeability ratios were calculated according to Goldman–Hodgkin–Katz relationship. For monovalent cations, permeability ratios were calculated as PX+/PK+ = exp(ΔVrevF/RT). For divalent cations, permeability ratios were calculated as: PX2+/PK+ = 𝛼K+[K + ]exp(ΔVrevF/RT)(1 + exp(ΔVrevF/RT))/ 4𝛼X2+[X2+] where Vrev is the reversal potential, F is Faraday’s constant, R is the universal gas constant, T is absolute temperature (where RT/F = 25.2 mV at room temperature) and 𝛼 is the ion activity coefficient (assumed to be 0.75 for K+ and 0.25 for Ca2+).
Mass spectrometry
Samples from 3a in DDM (before liposome incorporation), 3a in soy PC (1:50 ratio, and tested for activity in both electrophysiological and fluorescence-based assays) or control liposomes were run on SDS–PAGE gels and stained (Extended Data Fig. 9). For the DDM sample, the entire gel lane above the dye front was excised, including the stacking gel. For samples with lipid, to minimize lipid interference in sample digestion, the gel lane above the stained lipid was then excised (which includes the region around 15 kDa and larger).
Mass spectrometry was performed by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at UC Berkeley. The excised gel lanes were digested with trypsin in situ, and the resulting peptides extracted and concentrated. A nano liquid-chromatography column was packed in a 100-μm inner diameter glass capillary with an emitter tip. The column consisted of 10 cm of Polaris c18 5-μm packing material (Varian). The column was loaded by use of a pressure bomb and washed extensively with buffer A (5% acetonitrile/0.02% heptafluorobutyric acid). The column was then directly coupled to an electrospray ionization source mounted on a Thermo Fisher LTQ XL linear ion trap mass spectrometer. An Agilent 1200 HPLC equipped with a split line so as to deliver a flow rate of 300 nl min−1 was used for chromatography. Peptides were separated with a reverse phase elution using a gradient of 5 to 60% acetonitrile generated by a transition from buffer A to buffer B (60% acetonitrile/0.02% heptafluorobutyric acid) over 90 min.
Protein identification was done with Integrated Proteomics Pipeline (IP2, Integrated Proteomics Applications) using ProLuCID/Sequest, DTASelect2 and Census49,50,51. Tandem mass spectra were extracted into MS1 and MS2 files from raw files using RawExtractor52. Data were searched against a S. frugiperda protein database from Uniprot with the added amino acid sequence of purified 3a (including the cleavage site scar), supplemented with sequences of common contaminants and concatenated to form a decoy database53. Searches against soy (G. max) returned no hits for any sample. LTQ data were searched with 3000.0 milli-amu precursor tolerance and the fragment ions were restricted to a 600.0 ppm tolerance. All searches were parallelized and searched on the VJC proteomics cluster. Search space included all half tryptic peptide candidates with no missed cleavage restrictions. Carbamidomethylation (+57.02146) of cysteine was considered a static modification. To identify authentic termini, we required one tryptic terminus for each peptide identification. The ProLuCID search results were assembled and filtered using the DTASelect program with a peptide false discovery rate (FDR) of 0.001 for single peptides and a peptide FDR of 0.005 for additional peptides for the same protein. Under such filtering conditions, the estimated FDR was less than 1%.
Ca2+ uptake assay in proteoliposomes
Fluo-5N was incorporated into 3a-reconstituted proteoliposomes and control liposomes by first thawing frozen liposomes at a ratio of roughly 1:20 (v:v) into modified sucrose formation buffer54 25 mM HEPES pH 7.4, 150 mM KCl and 262 mM sucrose with a final concentration of 25 µM Fluo-5N (Stock concentration 5 mM in DMSO, AAT Bioquest). Next, the liposome mixtures in Eppendorf tubes were placed in a foam flotation in an ice bath and sonicated with a Branson Digital Sonifier 450 for 30 s in total (10% amplitude, 10 s pulse with 59-s wait time). Excess (unincorporated) Fluo-5N was then removed using microspin G-50 columns (Cytiva). The sample was then diluted into solution containing 50 mM HEPES, 300 mM KCl and 2 mM EGTA, pH 7.4, plated onto poly-d-lysine (Sigma-Aldrich, 1 mg ml−1) coated 96-well plates and centrifuged (440g for 5 min) at room temperature. Ca2+ influx was measured on addition of a 50 mM HEPES, 285 mM KCl, 10 mM CaCl2, pH 7.4 solution. Images were acquired every 3 s for a total of 150 s. Fluorescence intensity of each liposome was normalized to its average fluorescence before Ca2+ addition.
YO-PRO-1 flux assay
Liposomes were diluted in saline solution containing 300 mM KCl, 50 mM HEPES, pH 7.4 and plated onto poly-d-lysine (Sigma-Aldrich, 1 mg ml−1) coated 96-well plates, centrifuged (5,000g for 5 min) and incubated in 10 μM YO-PRO-1 iodide (Invitrogen) for 10 min at room temperature. Liposomes were rinsed with saline solution to remove free YO-PRO-1. Images were acquired using an ImageXpress Micro XLS microscope with a solid-state light source and ×20 air objective. Images were analyzed using MetaXpress 6 software (Molecular Devices). Empty liposomes displayed low-level fluorescence in the 488/540-nm range. Thus, liposomes were defined as YO-PRO-1+ if their fluorescence intensity was above the average intensity of unlabeled, empty liposomes. To calculate the total number of liposomes, we quantified the total number of liposomes using auto-segmentation on MetaXpress 6 from brightfield images. Stock solutions of ruthenium red (Tocris) were prepared in water, diluted in bath solution and applied to liposomes or cells 10 min before the start of the experiment. All statistical tests were performed using Prism (GraphPad). Values are reported as the mean ± s.d. or mean ± s.e.m as indicated. Mann–Whitney or a one-way or Welch’s analysis of variance (ANOVA) followed by the Dunnett’s, Sidak’s or Tukey’s post hoc tests (where appropriate) were used for statistical comparisons.
Nanodisc formation
Freshly purified 3a dimer in Buffer 1 was reconstituted into MSP1E3D1 nanodiscs with a mixture of lipids (DOPE:POPS:POPC at a 2:1:1 mass ratio, Avanti) at a final molar ratio of 1:4:400 (monomer ratio 3a, MSP1E3D1, lipid mixture). First, 20 mM solubilized lipid in Nanodisc formation buffer (20 mM HEPES, 150 mM KCl, 1 mM EDTA pH 7.4) was mixed with additional DDM detergent and 3a protein. This solution was mixed at 4 °C for 30 min before addition of purified MSP1E3D1. This addition brought the final concentrations to approximately 15 µM 3a, 60 µM MSP1E3D1, 6 mM lipid mix and 10 mM DDM in Nanodisc formation buffer. The solution with MSP1E3D1 was mixed at 4 °C for 10 min before addition of 200 mg of Biobeads SM2. Biobeads (washed into methanol, water, and then Nanodisc formation buffer) were weighed after liquid was removed by pipetting (damp weight). This mix was incubated at 4 °C for 30 min before addition of another 200 mg of Biobeads (for a total 400 mg of Biobeads per 0.5 ml of reaction). This final mixture was then gently tumbled at 4 °C overnight (roughly 12 h). Supernatant was cleared of beads by letting large beads settle and carefully removing liquid with a pipette. Sample was spun for 10 min at 21,000g before loading onto a Superdex 200 increase column in 20 mM HEPES, 150 mM KCl, pH 7.4. Peak fractions corresponding to 3a protein in MSP1E3D1 were collected, 10-kDa cutoff spin concentrated and used for grid preparation. MSP1E3D1 was prepared as described55 without cleavage of the His-tag. Tetrameric 3a in nanodiscs was prepared similarly, except with a ratio of 1:2:200 (monomer ratio 3a, MSP1E3D1, lipid mixture).
Grid preparation
Dimeric 3a in MSP1E3D1 was prepared at final concentration of 1.1 mg ml−1. For the sample with emodin (MillaporeSigma, cat. no. E7881), a stock solution of 50 mM emodin in DMSO added to protein sample for final concentrations of 1.1 mg ml−1 3a and 100 µM emodin and 1% DMSO. Concentrated sample was cleared by a 10 min 21,000g spin at 4 °C before grid making. For freezing grids, a 3-µl drop of protein was applied to freshly glow discharged Holey Carbon, 300 mesh R 1.2/1.3 gold grids (Quantifoil). A FEI Vitrobot Mark IV (Thermo Fisher Scientific) was used with 4 °C, 100% humidity, 1 blot force, a wait time of around 5 s and a 3 s blot time, before plunge freezing in liquid ethane. Grids were then clipped and used for data collection. Tetrameric 3a in MSP1E3D1 was frozen at 0.7 mg ml−1 with the same grid preparation. Unimaged grids from the same session were then shipped to the Netherlands for data collection on the 300 kV microscope with Falcon 4/CFEG/Selectris energy filter.
Cryo-EM data acquisition
For data collected on the Talos Arctica, grids were clipped and transferred to the microscope operated at 200 kV. Fifty frame videos were recorded on a Gatan K3 Summit direct electron detector in super-resolution counting mode with pixel size of 0.5685 Å. For the apo 3a dataset, the electron dose was 9.528 and 10.135 e− Å2 s−1 and total dose was 50.02 and 53.72 e− Å2 in the first set (1–2,007) and second set (2,008–6,309) of videos, respectively. The two different doses are the result of needing to restart the electron gun during collection. For the 3a with added emodin dataset, the electron dose was 8.991 e− Å2 s−1 and total dose was 47.21 e− Å2. For the 3a tetramer, the electron dose was 8.841 e− Å2 s−1 and total dose was 49.95 e− Å2. Nine videos were collected around a central hole position with image shift and defocus was varied from −0.6 to −2.0 µm through SerialEM56.
For data collected for the high-resolution 3a dimer structure, clipped grids from the same batch used on Talos Artica were sent to Thermo Fisher Scientific RnD division in Eindhoven, Netherlands. Grids were loaded onto the Krios G4 microscope equipped cold field emission gun (CFEG) operated at 300 kV. Data were collected on a Falcon 4 detector that was mounted behind a Selectris X energy filter. The slit width of the energy filter was set to 10 eV. Then 5,599 video stacks containing 1,429 raw frames were collected with the electron event representation (EER) mode57 of Falcon 4 detector at a magnification of ×165,000 corresponding to a pixel size of 0.727 Å. Each video stack was recorded with an exposure time of 6 s with a total dose of 50 e− /Å2 on sample and a defocus range between 0.5 and 1.2 µm.
Also see Table 1 for detailed data collection statistics.
Cryo-EM data processing
For the apo 3a dimer, motion correction and dose weighting were performed on all 6,309 videos using RELION 3.1’s implementation of MotionCor2, and twice ‘binned’ to 1.137 Å per pixel58,59,60. CTFFIND-4.1 was used to estimate the contrast transfer function (CTF) parameters61. Micrographs were then manually sorted to eliminate subjectively bad micrographs, such as empty or contaminated holes, resulting in 3,611 good micrographs. Additionally, micrographs with a CTF maximum resolution lower than 4 Å were discarded, resulting in 2,595 remaining micrographs. Template-free auto-picking of particles was performed with RELION3.1’s Laplacian-of-Gaussian filter yielding an initial set of particles. This initial set of particles were iteratively classified to generate templates, which were subsequently used to template-based auto-pick 1,750,730 particles.
Template-picked particles were iteratively two-dimensionally classified in RELION3.1 and then in cryoSPARC v.2 (ref. 62), resulting in 820,543 particles. These particles were subsequently three-dimensionally classified in cryoSPARC v.2 with iterative ab initio and heterogeneous refinement jobs. The resulting maps were visually evaluated with regard to the transmembrane domain density. A set of 86,479 particles were identified, polished in RELION3.1 and refined in cryoSPARC v.2 with subsequent homogeneous and nonuniform refinement63 jobs (maps were low-pass filtered to an initial resolution where TM density was still visible (6–9 Å), and the dynamic mask was tightened with the near (2–5 Å) and far (3–9 Å) parameters), yielding a map with overall resolution of 3.6 Å. UCSF pyem tools were used to convert data from cryoSPARC to RELION format64.
From this set of 86,479 particles, 2D classification was performed in RELION3.1 to identify a set of particles with subjectively equal view distribution. From the resulting set, 1,000 particles were randomly sampled and their coordinates used for training in the Topaz particle-picking pipeline65. Training, picking and extraction were performed independently on each subset of the micrographs. Then, 4,134,279 total particles were extracted in RELION3.1 with a box size of 256 pixels and ‘binned’ four times to 4.548 Å per pixel. These particles were then iteratively two-dimensionally classified in RELION3.1 resulting in 2,674,606 particles that were extracted at 2.274 Å per pixel. 2D classification was continued in both RELION3.1 and cryoSPARC v.2 resulting in 1,429,763 particles. Further classification was performed in cryoSPARC v.2 with subsequent ab initio (four classes, maximum resolution 8 Å) and heterogeneous refinement (8 Å initial resolution) jobs. The two best classes were selected and the particles pooled resulting in 743,800 particles, which were extracted in RELION3.1 at 1.137 Å per pixel.
Iterative three-dimensional (3D) classification was performed with subsequent ab initio and heterogeneous refinement jobs as described above. Following each round, 2D classification jobs were used to ‘rescue’ good particles from the worst classes before the next round. After three rounds, a final 2D classification job was used to identify 112,502 particles, which were subsequently pooled with the previous 86,479 RELION3.1 template-picked particles, resulting in 185,871 particles after duplicates (within 100 Å) were removed with RELION3.1.
These particles were then refined with subsequent homogeneous and nonuniform refinement jobs resulting in a map with overall resolution of 3.4 Å. This map was postprocessed in RELION3.1 using a mask with a soft edge (5-pixel extension, 7-pixel soft edge), the output of which was used for Bayesian particle polishing in RELION3.1 (training and polishing were each performed independently on each subset of the micrographs). The resulting ‘shiny’ particles were then refined in cryoSPARC v.2 with subsequent homogenous refinement (one extra pass, 7 Å initial resolution) and nonuniform refinement (C2, 1 extra pass, 9 Å initial resolution) to yield a map with 2.9-Å overall resolution.
For the 3a dimer with added 100 µM emodin, initial processing was similar to the dimer without the added drug (Supplementary Fig. 3). As with the apo 3a dimer, the critical steps included Topaz particle picking, particle clean-up with cryoSPARC v.2 ab initio and heterogeneous refinement, nonuniform refinement with tightened masking and RELION3.1 Bayesian particle polishing. However, in contrast to the apo dataset, we observed a set of particles that were included in <4-Å reconstructions that had discontinuous transmembrane domain density. Removal of these particles with RELION3.1 3D classification without angular sampling led to the best map from the emodin-added dataset. We did not see any evidence of bound emodin, but the 1% DMSO added with drug addition may have contributed to subtle map differences (Supplementary Fig. 3 and Extended Data Fig. 4).
For the 3a tetramer, the initial 7,092 micrographs were first cleaned using manual inspection and removal of images with <4 Å CtfMaxResolution to obtain a set of 4,324 micrographs. Reference particles for Topaz particle picking were generated by first template picking in RELION3.1, followed by 2D classification in both RELION3.1 and cryoSPARC v.2, and subsequent ab initio in cryoSPARC v.2. Particles from various views were then selected from iterative RELION3.1 2D classification to create a set of 6,843 particles. Using these coordinates for training, Topaz particle picking was then performed to generate a set of 1,282,913 initial particles. These particles were then cleaned using 2D classification in RELION3.1 and cryoSPARC v.2, followed by rounds of cryoSPARC v.2 ab initio and RELION3.1 3D classification. A major hurdle for tetramer processing was obtaining a reconstruction where most particles were properly oriented in the same direction (that is, intracellular domains on the same side of the nanodisc as seen in the 2D classes, Fig. 2f). Substantial, clean-up by 3D classification was needed to generate a correctly aligned reference map, but this map could then be used as a reference for refinements and classification for larger particle sets. Reconstructions with C1 or C2 symmetry looked similar (Extended Data Fig. 2e), although no tetramer reconstruction went to high enough resolution to determine symmetry with certainty. Therefore, it is possible that either the tetramer is pseudosymmetric or that different particles have heterogeneous orientations between dimer pairs. For the tetramer, the highest resolution reconstruction came from cryoSPARC v.2 nonuniform refinement with a tightened mask, which was subsequently used for dimer-docking and figure preparation.
For the apo 3a dimer imaged on a Krios with CFEG, Selectris X and Falcon 4, the EER video motion correction and subsequent polishing was performed in RELION3.1 using the devel-eer branch of Relion. Dose fractions consisting of 30 frames corresponding to 1.035 e− /Å2 per fraction were created. The initial 5,599 micrographs were pruned by selecting 4,495 micrographs with <3.5 Å CtfMaxResolution that also passed manual inspection. Similar to the Arctica datasets, an initial set of particles was generated with template-based picking and subsequent 2D classification, ab initio and heterogeneous refinement for clean-up was performed in cryoSPARC v.2 (Supplementary Fig. 4). This particle set (44,944) was then used as training for the first round of Topaz particle picking. For initial particle clean-up, the most important step was heterogeneous refinement in cryoSPARC v.2, yielding a particle set (215,227) that gave a C2 nonuniform refinement at 2.69 Å. For final particle clean-up, using RELION3.1, polished particles were subjected to 3D classification with no angular sampling and high tau (40) during which we monitored the rounds of classification by looking for convergence in resolution and inspecting maps to check for quality of protein features or noise. This type of job reliably allowed us to select the best particles (61,531) for high-resolution reconstruction at 2.26 Å.
At this point, a second Topaz training and picking round was conducted using this set of particles. After merging the best particles from both rounds of Topaz picking, removing potential duplicate particles within a distance of 100 Å and subsequent processing, we obtained a set of particles (91,218) that achieved a nonuniform reconstruction at 2.17 Å. Finally, rounds of CtfRefine in cryoSPARC v.2 and a final nonuniform refinement (with higher-order CTF terms enabled) achieved a map with an estimated resolution of 2.1 Å. Depending on inputs into sharpening and local resolution jobs (Extended Data Fig. 5a), the map has regions that give resolution estimates below 2 Å, consistent with our observations of holes in aromatic side chain density.
We note that the merging of two Topaz picked particle sets likely allowed us to break a particle-limited barrier to achieve the final reconstruction based on ResLog66 analysis performed in cryoSPARC (Extended Data Fig. 5c). Finally, we sampled various box sizes during processing (as large as 416 pixels and as small as 256 pixels)67. The best reconstructions were consistently achieved with our final box size of 300 pixels (218.1 Å).
Modeling, refinement and analysis
Apo dimeric 3a cryo-EM maps were sharpened using cryoSPARC and were of sufficient quality for de novo model building in Coot68. Real space refinement of the models was carried out using Phenix.real_space_refine69. Molprobity70 was used to evaluate the stereochemistry and geometry of the structure for subsequent rounds of manual adjustment in Coot and refinement in Phenix. For final sharpening and visualization of the high-resolution map we used Phenix Resolve Density Modification71. Docking of the apo dimeric 3a into the tetrameric 3a cryo-EM map was performed in Phenix using a map in which large empty regions of the nanodisc were erased in Chimera72. Similar results were found using maps with only the CDs present. Cavity measurements were made with HOLE73 implemented in Coot. Comparisons to the structure database was performed with DALI26. Structure prediction was performed with Phyre2 (ref. 74). Figures were prepared using PyMOL, Chimera, ChimeraX75, Fiji, Prism 8, GNU Image Manipulation Program and Adobe Photoshop and Illustrator software.
Fluorescence size-exclusion chromatography
Sf9 cells (roughly 4 million) from the third day of infection were pelleted, frozen and then thawed into extraction buffer (20 mM Tris pH 8, 150 mM KCl, all protease inhibitors used for protein purification, 1 mM EDTA, 1% DDM). Extraction was performed at 4 °C for 1 h and lysate was then pelleted at 21,000g at 4 °C for 1 h to clear supernatant. Supernatant was then run on a Superose 6 Increase column with fluorescence detection for GFP into 20 mM HEPES pH 7.4, 150 mM KCl and 0.025% DDM.
Transfection for confocal imaging
The constructs for full length 3a and 3aΔN were cloned into a vector with a CMV-promoter and C-terminal enhanced green fluorescent protein. Constructs (2 µg) were transfected into human embryonic kidney 293 cells on glass coverslips using Fugene HD (Promega) per the manufacturer’s instructions. Two days after transfection, cells were washed with DPBS and then fixed in 4% formaldehyde in DPBS for 10 min. Cells were then washed with DPBS before mounting the coverslip with Prolong Glass Antifade with NucBlue (Thermo Fisher Scientific) per the manufacturer’s instructions. Fluorescent images were collected using a Zeiss LSM 880 NLO AxioExaminer confocal microscope at either ×20 (numerical aperture 1.0) or ×63 oil immersion objective (numerical aperture 1.4). The samples were excited with 488 nm argon laser and we carried out image analysis using ImageJ (v.1.53a).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data associated with this study are publicly available. For cryo-EM of dimeric apo 3a, the final model is in the PDB under 6XDC, the final map is in the Electron Microscopy Data Bank (EMDB) under EMD-22136, and the original micrograph videos and final particle stack is in the Electron Microscopy Public Image Archive (EMPIAR) under EMPIAR-10439. For tetrameric apo 3a, the final map is in the EMDB under EMD-22138, and the original micrograph videos and final particle stack is in EMPIAR under EMPIAR-10441. For dimeric 3a in the presence of emodin, the final map is in the EMDB under EMD-22139, and the original micrograph videos and final particle stack is in EMPIAR under EMPIAR-10440. For the high-resolution dimeric apo 3a, the final model is in the PDB under 7KJR, the final map is in the EMDB under EMD-22898, and the original micrograph videos and final particle stack is in EMPIAR under EMPIAR-10612. For mass spectrometry searches, the S. frugiperda proteome was downloaded from Uniprot database (https://www.uniprot.org/uniprot/?query=taxonomy:7108). Source data are provided with this paper.
Change history
20 July 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41594-021-00642-1
References
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Walls, A. C. et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 183, 1735 (2020).
Shang, J. et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
Yin, W. et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499–1504 (2020).
Zhang, Y. & Kutateladze, T. G. Molecular structure analyses suggest strategies to therapeutically target SARS-CoV-2. Nat. Commun. 11, 2920–2924 (2020).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020).
Zhong, X. et al. Amino terminus of the SARS coronavirus protein 3a elicits strong, potentially protective humoral responses in infected patients. J. Gen. Virol. 87, 369–373 (2006).
Lu, B. et al. Humoral and cellular immune responses induced by 3a DNA vaccines against severe acute respiratory syndrome (SARS) or SARS-like coronavirus in mice. Clin. Vaccin. Immunol. 16, 73–77 (2009).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020).
Channappanavar, R., Fett, C., Zhao, J., Meyerholz, D. K. & Perlman, S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Virol. 88, 11034–11044 (2014).
Wang, H. et al. SARS-CoV-2 proteome microarray for mapping COVID-19 antibody interactions at amino acid resolution. ACS Cent. Sci. 6, 2238–2249 (2020).
Ward, D. et al. An integrated in silico immuno-genetic analytical platform provides insights into COVID-19 serological and vaccine targets. Genome Med 13, 4–12 (2021).
Siu, K.-L. et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 33, 8865–8877 (2019).
Chan, C.-M. et al. The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. Int. J. Biochem. Cell Biol. 41, 2232–2239 (2009).
Yue, Y. et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 9, 904–915 (2018).
Ren, Y. et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 17, 881–883 (2020).
Castaño-Rodriguez, C. et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. Mbio 9, 439 (2018).
Silvas, J. et al. Contribution of SARS-CoV-2 accessory proteins to viral pathogenicity in K18 hACE2 transgenic mice. Preprint at bioRxiv https://doi.org/10.1101/2021.03.09.434696 (2021).
Lu, W. et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl Acad. Sci. USA 103, 12540–12545 (2006).
Schwarz, S., Wang, K., Yu, W., Sun, B. & Schwarz, W. Emodin inhibits current through SARS-associated coronavirus 3a protein. Antivir. Res. 90, 64–69 (2011).
Delcour, A. H. (ed.) Electrophysiology of Unconventional Channels and Pores. (Springer International Publishing, 2015).
Nieva, J. L., Madan, V. & Carrasco, L. Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10, 563–574 (2012).
Scott, C. & Griffin, S. Viroporins: structure, function and potential as antiviral targets. J. Gen. Virol. 96, 2000–2027 (2015).
Nakane, T. et al. Single-particle cryo-EM at atomic resolution. Nature 587, 152–156 (2020).
Holm, L. & Sander, C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 (1995).
Dang, S. et al. Cryo-EM structures of the TMEM16A calcium-activated chloride channel. Nature 552, 426–429 (2017).
Paulino, C., Kalienkova, V., Lam, A. K. M., Neldner, Y. & Dutzler, R. Activation mechanism of the calcium-activated chloride channel TMEM16A revealed by cryo-EM. Nature 552, 421–425 (2017).
Zhang, M. et al. Structure of the mechanosensitive OSCA channels. Nat. Struct. Mol. Biol. 25, 850–858 (2018).
Jojoa-Cruz, S. et al. Cryo-EM structure of the mechanically activated ion channel OSCA1.2. Elife 7, e41845 (2018).
Chung, M.-K., Güler, A. D. & Caterina, M. J. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat. Neurosci. 11, 555–564 (2008).
Banke, T. G., Chaplan, S. R. & Wickenden, A. D. Dynamic changes in the TRPA1 selectivity filter lead to progressive but reversible pore dilation. Am. J. Physiol. Cell Physiol. 298, C1457–C1468 (2010).
Xu, L., Tripathy, A., Pasek, D. A. & Meissner, G. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J. Biol. Chem. 274, 32680–32691 (1999).
Choi, W., Clemente, N., Sun, W., Du, J. & Lu, W. The structures and gating mechanism of human calcium homeostasis modulator 2. Nature 576, 163–167 (2019).
Pope, L., Lolicato, M. & Minor, D. L. Polynuclear ruthenium amines inhibit K2P channels via a ‘finger in the dam’ mechanism. Cell Chem. Biol. 27, 511–524.e4 (2020).
Xie, L.-H., John, S. A. & Weiss, J. N. Spermine block of the strong inward rectifier potassium channel Kir2.1: dual roles of surface charge screening and pore block. J. Gen. Physiol. 120, 53–66 (2002).
Suma, A., Granata, D., Thomson, A. S., Carnevale, V. & Rothberg, B. S. Polyamine blockade and binding energetics in the MthK potassium channel. J. Gen. Physiol. 152, 6726 (2020).
Surprenant, A., Rassendren, F., Kawashima, E., North, R. A. & Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738 (1996).
Karasawa, A., Michalski, K., Mikhelzon, P. & Kawate, T. The P2X7 receptor forms a dye-permeable pore independent of its intracellular domain but dependent on membrane lipid composition. Elife 6, 3393 (2017).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Müller, M. A. et al. Human Coronavirus NL63 Open Reading Frame 3 encodes a virion-incorporated N-glycosylated membrane protein. Virol. J. 7, 6 (2010).
Wang, K. et al. PEDV ORF3 encodes an ion channel protein and regulates virus production. FEBS Lett. 586, 384–391 (2012).
Zhang, R. et al. The ORF4a protein of human coronavirus 229E functions as a viroporin that regulates viral production. Biochim. Biophys. Acta Biomembr. 1838, 1088–1095 (2014).
Miao, G. et al. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 56, 427–442.e5 (2020).
Ghosh, S. et al. β-coronaviruses use lysosomes for egress instead of the biosynthetic secretory pathway. Cell 183, 1520–1535.e14 (2020).
Zhao, J., Zhao, J. & Perlman, S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 84, 9318–9325 (2010).
Dietl, P., Haller, T. & Frick, M. Spatio-temporal aspects, pathways and actions of Ca2+ in surfactant secreting pulmonary alveolar type II pneumocytes. Cell Calcium 52, 296–302 (2012).
del Mármol, J., Rietmeijer, R. A. & Brohawn, S. G. in Potassium Channels: Methods and Protocols (eds Shyng, S.-L. et al.) 129–150 (Springer, 2018).
Xu, T. et al. ProLuCID: an improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteom. 129, 16–24 (2015).
Tabb, D. L., McDonald, W. H. & Yates, J. R. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 (2002).
Park, S. K., Venable, J. D., Xu, T. & Yates, J. R. A quantitative analysis software tool for mass spectrometry–based proteomics. Nat. Methods 5, 319–322 (2008).
McDonald, W. H. et al. MS1, MS2, and SQT—three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 18, 2162–2168 (2004).
Peng, J., Elias, J. E., Thoreen, C. C., Licklider, L. J. & Gygi, S. P. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC–MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 (2003).
Schöneberg, J. et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science 362, 1423–1428 (2018).
Ritchie, T. K. et al. Chapter 11—Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 (2009).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Guo, H. et al. Electron-event representation data enable efficient cryoEM file storage with full preservation of spatial and temporal resolution. IUCrJ 7, 860–869 (2020).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zivanov, J., Nakane, T. & Scheres, S. H. W. A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 163 (2018).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Asarnow, D., Palovcak, E. & Cheng, Y. asarnow/pyem: UCSF pyem v.0.5 (Zenodo, 2019); https://doi.org/10.5281/zenodo.3576630
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Stagg, S. M., Noble, A. J., Spilman, M. & Chapman, M. S. ResLog plots as an empirical metric of the quality of cryo-EM reconstructions. J. Struct. Biol. 185, 418–426 (2014).
Herzik, M. A., Wu, M. & Lander, G. C. High-resolution structure determination of sub-100 kDa complexes using conventional cryo-EM. Nat. Commun. 10, 1032–1039 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Terwilliger, T. C., Ludtke, S. J., Read, R. J., Adams, P. D. & Afonine, P. V. Improvement of cryo-EM maps by density modification. Nat. Methods 17, 923–927 (2020).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Smart, O. S., Neduvelil, J. G., Wang, X., Wallace, B. A. & Sansom, M. S. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph 14, 354–360 (1996).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
We thank H. Adesnik for providing emodin and for discussions. We thank P. Tobias for computational resources at the Cal-Cryo-EM facility and J. Hurley and E. Nogales for supporting the microscopy work. We thank Thermo Fisher Scientific for microscope access. We thank L. Kohlstaedt for assistance with mass spectrometry. We thank R. Rietmeijer for his gift of TRAAK proteoliposomes. We thank members of the Brohawn laboratory and A. Noble for thoughtful feedback on our preprint. Finally, we thank the many people at UC Berkeley and surrounding companies working during the pandemic who helped make this project possible. This work used the Vincent J Proteomics/Mass Spectrometry Laboratory at UC Berkeley, supported in part by NIH S10 Instrumentation grant no. S10RR025622. S.G.B. is a New York Stem Cell Foundation-Robertson Neuroscience Investigator. This work was funded by the New York Stem Cell Foundation; NIGMS grant no. GM123496; a McKnight Foundation Scholar Award; a Rose Hill Innovator Award; a Sloan Research Fellowship (to S.G.B.); NIGMS grant no. GM128263 (to D.M.K.); NSF Graduate Research Fellowship DGE1752814 (to S.S.M.); a Howard Hughes Medical Institute Faculty Scholar Award (to D.M.B.); and a Fast Grants Award from Emergent Ventures at the Mercatus Center, George Mason University (to D.M.B., S.G.B. and H. Adesnik).
Author information
Authors and Affiliations
Contributions
B.S., D.M.K., S.S.M., A.K., D.M.B. and S.G.B. conceived the project. D.M.K. performed all molecular biology, biochemistry, preparation of proteoliposomes and cryo-EM sample preparation. B.S. performed all electrophysiology. S.S.M. designed and performed the YO-PRO-1, ruthenium red blocker and Ca2+ flux assays with help from D.M.B. C.M.H. and D.M.K. processed the cryo-EM data. S.S. performed light microscopy. J.P.R. and D.B.T. collected cryo-EM data at UC Berkeley, and A.K. collected cryo-EM data at Thermo Fisher Scientific. S.G.B. built and refined the atomic models. D.M.B. and S.G.B. secured funding and supervised research. D.M.K., B.S., C.M.H., S.S.M., A.K., D.M.B. and S.G.B. wrote the paper with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
A.K. is an employee of Thermo Fisher Scientific. The other authors declare no competing interests.
Additional information
Peer review information Nature Structural & Molecular Biology thanks Toshimitsu Kawate, Vera Moiseenkova-Bell and Colin Nichols for their contribution to the peer review of this work. Peer reviewer reports are available. Florian Ullrich was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Purification and reconstitution of 3a.
a, Size exclusion chromatogram of 3a expressed in insect cells and extracted and purified in DDM (left). Pooled fractions corresponding to dimeric 3a are highlighted in blue. Coomassie-stained SDS-PAGE of pooled dimeric 3a-containing fractions (center) and of 3a following reconstitution into PC lipids (right). This is a representative preparation used for 3a proteoliposome experiments. WT 3a was purified into DDM six times with similar biochemical behavior. Proteoliposomes from three Soy PC preparations were used for activity assays. b, Size exclusion chromatogram of dimeric 3a reconstituted into MSP1E3D1 lipid nanodiscs (left). Pooled fractions are highlighted blue. 3a was incorporated into nanodiscs three separate times with similar biochemical behavior, and this representative preparation was used for dimeric 3a cryo-EM data collection (c,d) Same as (a,b), but for tetrameric 3a. The tetrameric peak has been observed in all WT 3a preparations. This representative preparation was used for cryo-EM of tetrameric 3a. e, GFP fluorescence chromatogram of 3a expressed in SF9 cells and extracted in DDM detergent. Samples were run on a Superose 6 column.
Extended Data Fig. 2 Example micrographs and 2D class averages of 3a structures.
a, Representative micrograph and (b) 2D class averages of dimeric apo 3a in MSP1E3D1 lipid nanodiscs. c, Representative micrograph and (d) 2D class averages of tetrameric apo 3a in MSP1E3D1 lipid nanodiscs. e, Map overview pipeline for final steps of tetramer processing (Also see Methods) (f) Representative micrograph and (g) 2D class averages of dimeric 3a in MSP1E3D1 lipid nanodiscs with emodin added. h, Representative micrograph and (i) 2D class averages of the Krios/CFEG/Selectris collected dimeric apo 3a in MSP1E3D1 lipid nanodiscs (from cryoSPARC). Micrograph scale bars, 100 Å.
Extended Data Fig. 3 Cryo-EM validation for dimeric apo 3a in MSP1E3D1 lipid nanodiscs.
a, Local resolution estimated in Relion colored as indicated on the final map. b, Angular distribution of particles used in final refinement with final map for reference. c, Fourier Shell Correlation (FSC) relationships (unmasked) between (black) the two unfiltered half-maps from refinement and used for calculating overall resolution at 0.143, (red) the final map and model, (gray) half-map one and model, and (blue) half-map and model.
Extended Data Fig. 4 Cryo-EM validation for dimeric 3a in MSP1E3D1 lipid nanodiscs with emodin added.
a, Local resolution estimated in Relion colored as indicated on the final map. b, Angular distribution of particles used in final refinement with final map for reference. c, Fourier Shell Correlation (FSC) relationships (masked) between (black) the two unfiltered half-maps from refinement and used for calculating overall resolution at 0.143, (red) the final map and model, (gray) half-map one and model, and (blue) half-map and model.
Extended Data Fig. 5 Cryo-EM validation for 2.1 Å dimeric apo 3a in MSP1E3D1 lipid nanodiscs.
a, Local resolution estimated in cryoSPARC at the indicated FSC thresholds colored as indicated on the density modified map. b, Angular distribution of particles used in final refinement with map for reference. c, ResLog analysis conducted in cryoSPARC at the indicated FSC thresholds. d, Fourier Shell Correlation as calculated in cryoSPARC before (top) and after (bottom) mask auto-tightening in the final round of refinement. e, Fourier Shell Correlation (FSC) relationships (masked) calculated in Phenix between (black) the two unfiltered half-maps from refinement and used for calculating overall resolution at 0.143, (red) the final map and model, (gray) half-map one and model, and (blue) half-map and model. f, Comparison of the original (2.9 Å) 3a map left to the high-resolution (2.1 Å) map before (middle) and after (right) Phenix density modification.
Extended Data Fig. 6 The dimer-dimer interface and a cysteine rich pocket in 3a.
a, A slice through the tetramer map with docked dimer models to illustrate the rigid body fit of model to map. Left: Same threshold as in Fig. 2 (0.25). Right: At a lower threshold (0.21). b, Dimer model (left) with boxed region zoom-in (middle) and an alternate view (right) to show the cysteine−rich region of 3a. Distances (dotted lines) between the reduced cysteines are displayed. c, Left: Dimers docked into the tetramer map with the solvent excluded surface of 3a colored from hydrophilic (dark cyan) to hydrophobic (dark orange). Right: ‘Open book’ view of each dimer from the perspective of the other dimer in the tetrameric assembly. The hydrophilic groove between TM2 and TM3 is outlined.
Extended Data Fig. 7 Localization of 3a-EGFP and 3aΔN-EGFP expressed in HEK293 cells.
a, 3a-GFP fluorescence localization in HEK293 cells transfected with 3a-EGFP or 3aΔN-EGFP. Single plane and brightest-point projection are displayed for each. Scale bar, 10 µm. b, Side view and view from the extracellular/lumenal space for dimeric 3a cryo-EM density (gray) from the original (left) and high-resolution (right) maps with unmodeled extended density above the mouth of the pore that may correspond to the N-terminal regions colored in purple. A 3a dimer model is drawn orange (TMD) and pink (CD) inside the density. The position of the final modeled N-terminal residue (S40) is indicated. c, 3a-EGFP or 3aΔN-EGFP field of view with multiple cells imaged using a 20X objective. Scale bar, 20 µm. d, Additional images of cells imaged with the 63X objective with both single plane and brightest-point projections displayed. Scale bar, 10 µm.
Extended Data Fig. 8 Patch recordings from 3a-proteoliposomes.
(a-c,g) Representative current recordings from 3a-proteoliposome. Currents were recorded with the following protocol: Vhold = 0 mV, Vtest = -100 to 100 mV, ΔV = 20 mV, ttest = 1 sec. Voltages indicated were corrected after recording for liquid junction potential. a, 150 mM K+ (black) or 150 mM Na+ (pink) bath solution. b, 150 mM K+ (black) or 75 mM Ca2+ (orange) bath solution. c, K+ (black) or 150 mM NMDG+ (blue) bath solution. d, Ba2+ does not block 3a currents. Current-voltage relationship plotted from a recording in 150 mM Na+ (pink) or 150 mM Na+ with 50 mM Ba2+ (green) bath solution. e, Emodin does not block 3a currents. Current-voltage relationship plotted from a recording in 150 mM Na+ (pink) or 150 mM Na+ with 100 µM emodin (green) bath solution. f, pH sensitivity of 3a. Current-voltage relationship plotted from a recording in 150 mM K+ pH 7.2 (black) and 150 mM K+ pH 9.2 (green) bath solution. (inset) Fold activation at +100 mV at pH 9.2 (mean ± s.e.m., n = 3) or pH 5.2 compared to pH 7.2 (mean ± s.e.m., n = 3). g, Representative current recordings from mock reconstituted (empty) liposome patch. h, Gap-free current recording in symmetric 150 mM KCl held at −80 mV during bath solution exchange and washout of varying Gd3+ concentrations represented by vertical green bars. Source data for parts d, e, and f are available online.
Extended Data Fig. 9 Mass spectrometry analysis of 3a and control samples.
a, SDS-PAGE gels with 3a and mock liposome samples prior to (left) and post (right) gel slice extraction for mass spectrometry. Approximate outline of the extracted region is shown with a dotted line. The control and 3a samples were from a liposome preparation used for both electrophysiological and fluorescence assays. b, Same as (a), but for 3a in DDM sample (prior to liposome incorporation). This sample was from a DDM prep used for both cryo-EM and proteoliposome preparations. c, Summary table of mass spectrometry analysis for the three analyzed samples. The only other hits were to human keratin proteins from the common contaminant library. No additional hits were detected from the G. max (soy) search.
Extended Data Fig. 10 Purification and liposome reconstitution of 3a mutants and truncation.
Top: GFP fluorescence chromatogram (FSEC) of 3a, as well as indicated mutants and truncations, expressed in SF9 cells and extracted in DDM detergent. Samples were run on a Superose 6 column. For all other panels: Size exclusion chromatogram from a s200 column of indicated 3a constructs expressed in insect cells and extracted and purified in DDM (left), coomassie-stained SDS-PAGE of pooled dimeric 3a construct -containing fractions (center), and of 3a following reconstitution into PC lipids (right). These gels and chromatograms are from the preparations used for electrophysiological assays.
Supplementary information
Source data
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Rights and permissions
About this article

Cite this article
Kern, D.M., Sorum, B., Mali, S.S. et al. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat Struct Mol Biol 28, 573–582 (2021). https://doi.org/10.1038/s41594-021-00619-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-021-00619-0
This article is cited by
-
Lipid nanodisc scaffold and size alter the structure of a pentameric ligand-gated ion channel
Nature Communications (2024)
-
Molecular basis of the inositol deacylase PGAP1 involved in quality control of GPI-AP biogenesis
Nature Communications (2024)
-
SARS-CoV-2 biology and host interactions
Nature Reviews Microbiology (2024)
-
SARS-CoV-2 virulence factor ORF3a blocks lysosome function by modulating TBC1D5-dependent Rab7 GTPase cycle
Nature Communications (2024)
-
Bridging structural and cell biology with cryo-electron microscopy
Nature (2024)
