
Abstract
Synonymous codon choice can have dramatic effects on ribosome speed and protein expression. Ribosome profiling experiments have underscored that ribosomes do not move uniformly along mRNAs. Here, we have modeled this variation in translation elongation by using a feed-forward neural network to predict the ribosome density at each codon as a function of its sequence neighborhood. Our approach revealed sequence features affecting translation elongation and characterized large technical biases in ribosome profiling. We applied our model to design synonymous variants of a fluorescent protein spanning the range of translation speeds predicted with our model. Levels of the fluorescent protein in budding yeast closely tracked the predicted translation speeds across their full range. We therefore demonstrate that our model captures information determining translation dynamics in vivo; that this information can be harnessed to design coding sequences; and that control of translation elongation alone is sufficient to produce large quantitative differences in protein output.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Change history
09 July 2018
In the version of this article initially published, Supplementary Notes 1—3 and Supplementary Tables 1 and 2 were omitted from the Supplementary Text and Figures file. The error has been corrected.
References
Ishimura, R. et al. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459 (2014).
Goodarzi, H. et al. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165, 1416–1427 (2016).
Kirchner, S. et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 15, e2000779 (2017).
Zhao, F., Yu, C.-H. & Liu, Y. Codon usage regulates protein structure and function by affecting translation elongation speed in Drosophila cells. Nucleic Acids Res. 45, 8484–8492 (2017).
Shah, P., Ding, Y., Niemczyk, M., Kudla, G. & Plotkin, J. B. Rate-limiting steps in yeast protein translation. Cell 153, 1589–1601 (2013).
Presnyak, V. et al. Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124 (2015).
Bazzini, A. A. et al. Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. EMBO J. 35, 2087–2103 (2016).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. S. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Stadler, M. & Fire, A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17, 2063–2073 (2011).
Dana, A. & Tuller, T. Determinants of translation elongation speed and ribosomal profiling biases in mouse embryonic stem cells. PLOS Comput. Biol. 8, e1002755 (2012).
Charneski, C. A. & Hurst, L. D. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 11, e1001508 (2013).
Gardin, J. et al. Measurement of average decoding rates of the 61 sense codons in vivo. eLife 3, e03735 (2014).
Lareau, L. F., Hite, D. H., Hogan, G. J. & Brown, P. O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife 3, e01257 (2014).
Pop, C. et al. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol. Syst. Biol. 10, 770 (2014).
Liu, T.-Y. & Song, Y. S. Prediction of ribosome footprint profile shapes from transcript sequences. Bioinformatics 32, i183–i191 (2016).
Zhang, S. et al. Analysis of ribosome stalling and translation elongation dynamics by deep learning. Cell Syst. 5, 212–220.e6 (2017).
Dao Duc, K. & Song, Y. S. The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLoS Genet. 14, e1007166 (2018).
Fang, H. et al. Scikit-ribo enables accurate estimation and robust modeling of translation dynamics at codon resolution. Cell Syst. 6, 180–191.e4 (2018).
O’Connor, P. B. F., Andreev, D. E. & Baranov, P. V. Comparative survey of the relative impact of mRNA features on local ribosome profiling read density. Nat. Commun. 7, 12915 (2016).
Artieri, C. G. & Fraser, H. B. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res. 24, 2011–2021 (2014).
Hussmann, J. A., Patchett, S., Johnson, A., Sawyer, S. & Press, W. H. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet. 11, e1005732 (2015).
Weinberg, D. E. et al. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 14, 1787–1799 (2016).
Sharp, P. M., Tuohy, T. M. & Mosurski, K. R. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14, 5125–5143 (1986).
Plotkin, J. B. & Kudla, G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 (2011).
dos Reis, M., Savva, R. & Wernisch, L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044 (2004).
Letzring, D. P., Dean, K. M. & Grayhack, E. J. Control of translation efficiency in yeast by codon-anticodon interactions. RNA 16, 2516–2528 (2010).
Gamble, C. E., Brule, C. E., Dean, K. M., Fields, S. & Grayhack, E. J. Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166, 679–690 (2016).
Murphy, F. V. IV & Ramakrishnan, V. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol. 11, 1251–1252 (2004).
McGlincy, N. J. & Ingolia, N. T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129 (2017).
Iwasaki, S., Floor, S. N. & Ingolia, N. T. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature 534, 558–561 (2016).
Schuller, A. P., Wu, C. C.-C., Dever, T. E., Buskirk, A. R. & Green, R. eIF5A functions globally in translation elongation and termination. Mol. Cell 66, 194–205.e5 (2017).
Sheff, M. A. & Thorn, K. S. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21, 661–670 (2004).
Chu, D. et al. Translation elongation can control translation initiation on eukaryotic mRNAs. EMBO J. 33, 21–34 (2014).
Qian, W., Yang, J.-R., Pearson, N. M., Maclean, C. & Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 8, e1002603 (2012).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Battenberg, E. et al. Lasagne: first release. https://doi.org/10.5281/zenodo.27878 (2015).
The Theano Development Team et al. Theano: a Python framework for fast computation of mathematical expressions. https://arxiv.org/abs/1605.02688 (2016).
Ritz, C. & Spiess, A.-N. qpcR: an R package for sigmoidal model selection in quantitative real-time polymerase chain reaction analysis. Bioinformatics 24, 1549–1551 (2008).
Burdukiewicz, M. et al. Methods for comparing multiple digital PCR experiments. Biomol. Detect. Quantif. 9, 14–19 (2016).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Gueldener, U., Heinisch, J., Koehler, G. J., Voss, D. & Hegemann, J. H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23 (2002).
Daniel Gietz, R. & Woods, R. A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 (2002).
Hahne, F. et al. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 10, 106 (2009).
Sarkar, D., Le Meur, N. & Gentleman, R. Using flowViz to visualize flow cytometry data. Bioinformatics 24, 878–879 (2008).
Ares, M. Isolation of total RNA from yeast cell cultures. Cold Spring Harb. Protoc. 2012, 1082–1086 (2012).
Acknowledgements
We are grateful to N. Ingolia and S. McCurdy for discussion. This work was supported by the National Cancer Institute of the National Institutes of Health, under award R21CA202960 to L.F.L., and by the National Institute of General Medical Sciences of the National Institutes of Health, under award P50GM102706 to the Berkeley Center for RNA Systems Biology. R.T. was supported by the Department of Defense through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. This work made use of the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley, supported by National Institutes of Health S10 Instrumentation grant OD018174, and the UC Berkeley flow cytometry core facilities.
Author information
Authors and Affiliations
Contributions
L.F.L., R.T., and N.J.M. designed the study, with input from L.P. R.T. developed the software and performed modeling, and R.T., L.P., and L.F.L. analyzed and interpreted the computational results. N.J.M. designed and created the yeast strains and performed expression experiments, with assistance from M.E.G. and N.N. M.E.G. performed yeast ribosome profiling. N.J.M. and L.F.L. analyzed and interpreted the experimental data. R.T. and L.F.L. wrote the manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Neural network model structure.
(A) Counts are predicted at the A site codon. In the model shown, a sequence neighborhood spanning from 5 codons upstream of the A site (codon -5) to 4 codons downstream of the A site (codon + 4) is used as the predictive region. This neighborhood is divided into codons and encoded via one-hot encoding (purple) for input into a regression model. We also encode the same region as nucleotide features (green) and include these features in the model. Finally, we compute RNA structure scores on three 30 nt sliding structure windows that span the width of a typical 28 nt footprint. These windows start 17, 16, and 15 nucleotides before the start of the A site. (B) These features are concatenated in a vector, which is used as the input to a fully connected feedforward neural network model. Each model in this paper contains one hidden layer with 200 hidden units, and a tanh activation function on the hidden units. The output layer contains one unit with a ReLU activation function to enforce nonnegativity of predicted scaled counts.
Supplementary Figure 2 Change in MSE upon including mRNA structure.
We compared the performance of a model using a sequence neighborhood from codons -7 to + 5 against models that each also include the folding energy of a single 30 nt window. Each point shows the change in MSE upon including a particular window. The greatest improvement in MSE is achieved by including a window starting at nucleotide position -17 and ending at position 12. This is roughly coterminal with a typical 28 nt ribosome footprint (nucleotide positions -15 to + 12).
Supplementary Figure 3 Relative contributions of A-site codons and context.
(A) Pearson correlation of observed vs. predicted scaled counts per codon, for a model using codons -3 to + 2 and associated nucleotides (x-axis) and a model using the same region but without the A site (y-axis). Codons whose inclusion in the model leads to significantly better prediction (higher correlation between the observed and predicted scaled counts), per a t-test of the Fisher transformation of correlations with an FDR of 5%, are shown in red. (B) We extracted the sequence context (codons -3 to + 2) for all positions where the squared error was higher for an A-site-only model than for a model with codons -3 to + 2 but no A site. The proportion of codons at each position in this set was compared to the overall distribution of codons in the test set with a two-sided proportion test, using an FDR of 5%, and codons with adjusted p < 0.05 are shown in the plot.
Supplementary Figure 4 Predictive values of codon positions.
Predictive value of codon positions in (A) a human ribosome profiling data set using Circligase II (Iwasaki, S. et al., Nature. 534(7608), 558-61, 2016), and (B) a yeast ribosome profiling dataset using Circligase I (Schuller, A. et al., Mol. Cell. 66, 194–205.e5, 2017). As in Fig. 3a, we trained a reference model on codons -7 to + 5 (with nucleotide features over the same neighborhood), and then a series of leave-one-out models each excluding exactly one codon in the sequence neighborhood, along with the corresponding nucleotides. For each model, we compute Pearson correlations between the true and predicted scaled counts over all codons in the test set. Shown is the difference in Pearson correlations between the reference model and the leave-one-out models. Higher Δr indicates increased importance of that codon position to model predictions.
Supplementary Figure 5 Circligase I and II preferences.
(A) Mean contributions to scaled counts at the 5´ end of a ribosome footprint, for yeast data sets generated with Circligase II (our data) and Circligase I (Schuller, A. et al., Mol. Cell. 66, 194–205.e5, 2017). Scores are from the -5 codon position. To generate these scores, we trained models only on 28 nt footprints with their 5´ end aligning with the beginning of the -5 codon. (B) Ligation efficiency of CircLigase I enzyme, as in Fig. 3g.
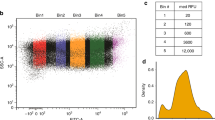
Supplementary Figure 6 Flow cytometry gating strategy.
Scatter plot of forward scatter area (FSC, arbitrary units) against side scatter area (SSC, arbitrary units) for each of the 50000 events collected for a representative flow cytometry sample of diploid yeast expressing mCherry and a differentially optimized eCitrine. Events are colored by their density on the plot, low density points being colored blue moving to high density events being colored dark red. Events outside the plotted area are denoted by grey lines at the edge of the plot. Annotated by red regions are seven areas of high local density defined by the curv2filter method, each with the number of events they contain. For each sample, events within the most populous region were taken forward for further analysis; in this representative sample, these would be the events within Area 3.
Supplementary Figure 7 yECitrine mRNA level and fluorescence.
(A) eCitrine:mCherry fluorescence ratio as in Fig. 3c, including this ratio for the yECitrine sequence (magenta). (B) eCitrine:mCherry mRNA ratio measured by qPCR in biological replicates of four strains (colors as in Fig. 4). Each data point represents the ratio of medians of three technical replicates, normalized to the median ratio of the highest expression strain.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Notes 1–3 and Supplementary Tables 1 and 2
Supplementary Table 3
Codon contributions. Weights learned as described in the methods for each codon in positions -5 to +4, used to create figure 3B
Supplementary Dataset 1
Annotated sequences of plasmids for eCitrine strain creation, in genbank format
Rights and permissions
About this article

Cite this article
Tunney, R., McGlincy, N.J., Graham, M.E. et al. Accurate design of translational output by a neural network model of ribosome distribution. Nat Struct Mol Biol 25, 577–582 (2018). https://doi.org/10.1038/s41594-018-0080-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-018-0080-2
This article is cited by
-
Riboformer: a deep learning framework for predicting context-dependent translation dynamics
Nature Communications (2024)
-
Using protein-per-mRNA differences among human tissues in codon optimization
Genome Biology (2023)
-
RPL3L-containing ribosomes determine translation elongation dynamics required for cardiac function
Nature Communications (2023)
-
Streamlined and sensitive mono- and di-ribosome profiling in yeast and human cells
Nature Methods (2023)
-
Molecular engineering improves antigen quality and enables integrated manufacturing of a trivalent subunit vaccine candidate for rotavirus
Microbial Cell Factories (2021)
