
Abstract
N-linked glycosylation is a predominant post-translational modification of protein in eukaryotes, and its dysregulation is the etiology of several human disorders. The enzyme UDP-N-acetylglucosamine:dolichyl-phosphate N-acetylglucosaminephosphotransferase (GlcNAc-1-P-transferase or GPT) catalyzes the first and committed step of N-linked glycosylation in the endoplasmic reticulum membrane, and it is the target of the natural product tunicamycin. Tunicamycin has potent antibacterial activity, inhibiting the bacterial cell wall synthesis enzyme MraY, but its usefulness as an antibiotic is limited by off-target inhibition of human GPT. Our understanding of how tunicamycin inhibits N-linked glycosylation and efforts to selectively target MraY are hampered by a lack of structural information. Here we present crystal structures of human GPT in complex with tunicamycin. Structural and functional analyses reveal the difference between GPT and MraY in their mechanisms of inhibition by tunicamycin. We demonstrate that this difference could be exploited to design MraY-specific inhibitors as potential antibiotics.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Lehrman, M. A. Biosynthesis of N-acetylglucosamine-P-P-dolichol, the committed step of asparagine-linked oligosaccharide assembly. Glycobiology 1, 553–562 (1991).
Belaya, K. et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet 91, 193–201 (2012).
Wu, X. et al. Deficiency of UDP-GlcNAc:dolichol phosphate N-acetylglucosamine-1 phosphate transferase (DPAGT1) causes a novel congenital disorder of glycosylation type Ij. Hum. Mutat. 22, 144–150 (2003).
Würde, A. E. et al. Congenital disorder of glycosylation type Ij (CDG-Ij, DPAGT1-CDG): extending the clinical and molecular spectrum of a rare disease. Mol. Genet. Metab. 105, 634–641 (2012).
Liwosz, A., Lei, T. & Kukuruzinska, M. A. N-glycosylation affects the molecular organization and stability of E-cadherin junctions. J. Biol. Chem. 281, 23138–23149 (2006).
Nita-Lazar, M. et al. Overexpression of DPAGT1 leads to aberrant N-glycosylation of E-cadherin and cellular discohesion in oral cancer. Cancer Res. 69, 5673–5680 (2009).
Lehrman, M. A. A family of UDP-GlcNAc/MurNAc: polyisoprenol-P GlcNAc/MurNAc-1-P transferases. Glycobiology 4, 768–771 (1994).
Takatsuki, A., Arima, K. & Tamura, G. Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. J. Antibiot. (Tokyo) 24, 215–223 (1971).
Takatsuki, A. et al. The structure of tunicamycin. Agric. Biol. Chem. 41, 2307–2309 (1977).
Wang, R. et al. A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg. Med. Chem. 5, 661–672 (1997).
Izumi, M., Yuasa, H. & Hashimoto, H. Bisubstrate analogues as glycosyltransferase inhibitors. Curr. Top. Med. Chem. 9, 87–105 (2009).
Price, N. P. & Momany, F. A. Modeling bacterial UDP-HexNAc: polyprenol-P HexNAc-1-P transferases. Glycobiology 15, 29R–42R (2005).
Xu, L., Appell, M., Kennedy, S., Momany, F. A. & Price, N. P. Conformational analysis of chirally deuterated tunicamycin as an active site probe of UDP-N-acetylhexosamine:polyprenol-P N-acetylhexosamine-1-P translocases. Biochemistry 43, 13248–13255 (2004).
Elbein, A. D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu. Rev. Biochem. 56, 497–534 (1987).
Keller, R. K., Boon, D. Y. & Crum, F. C. N-Acetylglucosamine-1-phosphate transferase from hen oviduct: solubilization, characterization, and inhibition by tunicamycin. Biochemistry 18, 3946–3952 (1979).
Lehle, L. & Tanner, W. The specific site of tunicamycin inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 71, 167–170 (1976).
Takatsuki, A., Kohno, K. & Tamura, G. Inhibition of biosynthesis of polyisoprenol sugars in chick embryo microsomes by tunicamycin. Agric. Biol. Chem. 39, 2089–2091 (1975).
Tkacz, J. S. & Lampen, O. Tunicamycin inhibition of polyisoprenyl N-acetylglucosaminyl pyrophosphate formation in calf-liver microsomes. Biochem. Biophys. Res. Commun. 65, 248–257 (1975).
Duksin, D. & Mahoney, W. C. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J. Biol. Chem. 257, 3105–3109 (1982).
Faye, L. & Chrispeels, M. J. Apparent inhibition of beta-fructosidase secretion by tunicamycin may be explained by breakdown of the unglycosylated protein during secretion. Plant Physiol. 89, 845–851 (1989).
Koizumi, N., Ujino, T., Sano, H. & Chrispeels, M. J. Overexpression of a gene that encodes the first enzyme in the biosynthesis of asparagine-linked glycans makes plants resistant to tunicamycin and obviates the tunicamycin-induced unfolded protein response. Plant Physiol. 121, 353–361 (1999).
Oslowski, C. M. & Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92 (2011).
Brandish, P. E. et al. Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: inhibition of phospho-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob. Agents Chemother. 40, 1640–1644 (1996).
Tamura, G., Sasaki, T., Matsuhashi, M., Takatsuki, A. & Yamasaki, M. Tunicamycin inhibits the formation of lipid intermediate in cell-free peptidoglycan synthesis of bacteria. Agric. Biol. Chem. 40, 447–449 (1976).
Lukose, V., Walvoort, M. T. C. & Imperiali, B. Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface. Glycobiology 27, 820–833 (2017).
Chung, B. C. et al. Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 341, 1012–1016 (2013).
Chung, B. C. et al. Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature 533, 557–560 (2016).
Hakulinen, J. K. et al. MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat. Chem. Biol. 13, 265–267 (2017).
Al-Dabbagh, B. et al. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 47, 8919–8928 (2008).
Bouhss, A., Trunkfield, A. E., Bugg, T. D. & Mengin-Lecreulx, D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 32, 208–233 (2008).
Dan, N. & Lehrman, M. A. Oligomerization of hamster UDP-GlcNAc:dolichol-P GlcNAc-1-P transferase, an enzyme with multiple transmembrane spans. J. Biol. Chem. 272, 14214–14219 (1997).
Gupta, K. et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017).
Plouhar, P. L. & Bretthauer, R. K. A phospholipid requirement for dolichol pyrophosphate N-acetylglucosamine synthesis in phospholipase A2-treated rat lung microsomes. J. Biol. Chem. 257, 8907–8911 (1982).
Plouhar, P. L. & Bretthauer, R. K. Restoration by phospholipids of dolichol pyrophosphate N-acetylglucosamine synthesis in delipidated rat lung microsomes. J. Biol. Chem. 258, 12988–12993 (1983).
Kaushal, G. P. & Elbein, A. D. Purification and properties of UDP-GlcNAc:dolichyl-phosphate GlcNAc-1-phosphate transferase. Activation and inhibition of the enzyme. J. Biol. Chem. 260, 16303–16309 (1985).
Walvoort, M. T., Lukose, V. & Imperiali, B. A modular approach to phosphoglycosyltransferase inhibitors inspired by nucleoside antibiotics. Chemistry 22, 3856–3864 (2016).
Yamamoto, K., Yakushiji, F., Matsumaru, T. & Ichikawa, S. Total synthesis of tunicamycin V. Org. Lett. 20, 256–259 (2018).
Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 (2010).
Foadi, J. et al. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 69, 1617–1632 (2013).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 (2011).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Tickle, I. J. et al. STARANISO. (Global Phasing Ltd, 2017).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Kovalevskiy, O., Nicholls, R. A. & Murshudov, G. N. Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr. D Struct. Biol. 72, 1149–1161 (2016).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Delano, W. L. The PyMol Molecular Graphics System. (DeLano Scientific, 2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Celniker, G. et al. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 53, 199–206 (2013).
Bouhss, A., Crouvoisier, M., Blanot, D. & Mengin-Lecreulx, D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 279, 29974–29980 (2004).
Acknowledgements
Data for this study were collected at beamlines NECAT 24-ID-C and 24-ID-E and at SERCAT 22-ID, both at the Advanced Photon Source. We thank B. Chung for initial GPT biochemistry. We thank I. Tickle and the STARANISO team for help with our anisotropic data analysis. This work was supported by the National Institutes of Health (R01GM120594 and R35NS097241 to S.-Y. L.), JSPS Grant-in-Aid for Scientific Research (B) (16H05097 to S.I.), Astellas Foundation for Research on Metabolic Disorders (to S.I.), Hokkaido University GFC, PSOU, funded by MEXT (to S.I.), and BINDS from the Japan Agency for Medical Research and Development (to S.I.). Beamlines 24-ID-C and 24-ID-E are funded by P41 GM103403 and S10 RR029205.
Author information
Authors and Affiliations
Contributions
J.Y. performed GPT crystallization, data collection, and protein preparation for functional studies; E.H.M. performed all functional studies of GPT and MraY; B.K. assisted in sample purification for functional studies; A.C.Y.K. and J.Y. performed X-ray data processing, model building and refinement. J.Y., A.C.Y.K., B.K., and E.H.M. are under the guidance of S.-Y.L. S.I. designed tunicamycin-MurNAc. K.Y. synthesized the tunicamycin-MurNAc under the guidance of S.I. S.-Y.L., A.C.Y.K., E.H.M., and J.Y. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Chemical structures of tunicamycin and the natural substrates of GPT and MraY.
Chemical structure of the competitive inhibitor tunicamycin A1 is shown at the top, while the structures of the natural substrates (UDP-GlcNAc for GPT and UDP-MurNAc-pentapeptide for MraY) are shown at the bottom. All compounds contain uridine moieties and either a GlcNAc or MurNAc moiety.
Supplementary Figure 2 The His129 and Pro129 variants of hGPT are active and have comparable specific activity.
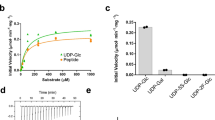
a, TLC plate image of hGPT His129- and Pro129-catalyzed reactions (in triplicate) containing 200 nM of each enzyme, 500 μM C55-dol-P, 0.1 mM UDP-GlcNAc and 0.01 mM [14C]UDP-GlcNAc, 70 mM Tris-HCl pH 8.0, 500 mM NaCl, 80 mM MgCl2, 5 mM DM, 1 mg/mL POPG, and 10% glycerol. A time course assay was conducted at 30°C and 2 μL of each reaction was spotted on the TLC plate every 5 min; the 15 min time point shown is within the linear range. b, Specific activity measurements based on spot intensity quantification of the substrate and product bands shown in panel a. Three technical replicates are shown, with the mean value represented by a line.
Supplementary Figure 3 Composite omit electron density of hGPT.
2F o −F c composite omit maps were calculated from the canonical hGPT Pro129 data (3.1 Å resolution) omitting 5% of the model at a time, and contoured to 0.8 σ. a, hGPT dimer, showing ribbons for protomer A only. Omit density for protomer B is colored pink for clarity. b, View of the cytosolic cavity, showing tunicamycin and several nearby residues. c, View of the dimer interface from within the membrane, looking at protomer A from protomer B. d, View of the dimer interface from the cytosol. e, 2F o −F c composite omit map, shown for individual transmembrane helices 1 to 10 and carved 1.8 Å from the model.
Supplementary Figure 4 Stereo views of tunicamycin bound to hGPT.
The canonical hGPT (Pro129) is shown at the top (3.1 Å resolution), while the His129 variant is shown below (2.95 Å resolution). Tunicamycin is denoted as magenta sticks. 2F o –F c omit density of tunicamycin is shown in blue mesh (contoured to 0.8 σ), while the F o –F c omit density of tunicamycin is shown in green mesh (contoured to 3 σ). Maps were carved 1.8 Å from the tunicamycin model.
Supplementary Figure 5 Alignment of GPT sequences.
The sequences of human GPT, MraYAA (5CKR), and MraYCB (5JNQ) were first aligned by structural superposition, expanded to 45 GPT ortholog sequences by MAFFT, and corrected manually. Invariant positions (100% identity) are shaded red, while conserved positions (>90% identity) are shaded gray. The secondary structure of human GPT is shown, highlighting the insertion in loop E that is absent in MraY sequences (TM, transmembrane α-helix; α, α-helix; β, β-strand).
Supplementary Figure 6 Omit density of POPG lipid molecules.
Omit maps were calculated with data of the canonical hGPT (Pro129) to 3.1 Å resolution. One lipid tail of 1-palmitoyl-2-oleoylglycero-3-phosphoglycerol (POPG, magenta sticks) is found in each side fenestration. 2F o –F c omit density of POPG molecules is shown in blue mesh (contoured to 0.8 σ), while the F o –F c omit density is shown in green mesh (contoured to 3 σ). Maps were carved 1.8 Å from the POPG molecules.
Supplementary Figure 7 Mapping of human disease mutants to the structure of hGPT.
Positions of CDG1J-associated mutants are colored yellow, while positions of CMS13-associated mutants are colored blue.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7 and Supplementary Note
Rights and permissions
About this article

Cite this article
Yoo, J., Mashalidis, E.H., Kuk, A.C.Y. et al. GlcNAc-1-P-transferase–tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol 25, 217–224 (2018). https://doi.org/10.1038/s41594-018-0031-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-018-0031-y
This article is cited by
-
The tunicamycin derivative TunR2 exhibits potent antibiotic properties with low toxicity in an in vivo Mycobacterium marinum-zebrafish TB infection model
The Journal of Antibiotics (2024)
-
Establishment of a novel ER-stress induced myopia model in mice
Eye and Vision (2023)
-
Targeting N-glycosylation of 4F2hc mediated by glycosyltransferase B3GNT3 sensitizes ferroptosis of pancreatic ductal adenocarcinoma
Cell Death & Differentiation (2023)
-
DNMT2/TRDMT1 gene knockout compromises doxorubicin-induced unfolded protein response and sensitizes cancer cells to ER stress-induced apoptosis
Apoptosis (2023)
-
3-Ketodihydrosphingosine reductase maintains ER homeostasis and unfolded protein response in leukemia
Leukemia (2022)
