
Abstract
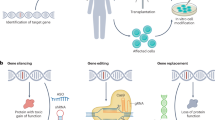
Gene therapy is making a comeback. With its twin promise of targeting disease etiology and ‘long-term correction’, gene-based therapies (defined here as all forms of genome manipulation) are particularly appealing for neurodegenerative diseases, for which conventional pharmacologic approaches have been largely disappointing. The recent success of a viral-vector-based gene therapy in spinal muscular atrophy—promoting survival and motor function with a single intravenous injection—offers a paradigm for such therapeutic intervention and a platform to build on. Although challenges remain, the newfound optimism largely stems from advances in the development of viral vectors that can diffusely deliver genes throughout the CNS, as well as genome-engineering tools that can manipulate disease pathways in ways that were previously impossible. Surely spinal muscular atrophy cannot be the only neurodegenerative disease amenable to gene therapy, and one can imagine a future in which the toolkit of a clinician will include gene-based therapeutics. The goal of this Review is to highlight advances in the development and application of gene-based therapies for neurodegenerative diseases and offer a prospective look into this emerging arena.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Somanathan, S., Calcedo, R. & Wilson, J. M. Adenovirus–antibody complexes contributed to lethal systemic inflammation in a gene therapy trial. Mol. Ther. 28, 784–793 (2020).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Keeler, C. E. Gene therapy. J. Hered. 38, 294–298 (1947).
Logovinsky, V. et al. Safety and tolerability of BAN2401—a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 8, 14 (2016).
Hudry, E. & Vandenberghe, L. H. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron 102, 839–862 (2019).
Doudna, J. A. The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020).
Huang, L. K., Chao, S. P. & Hu, C. J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27, 18 (2020).
Bedbrook, C. N., Deverman, B. E. & Gradinaru, V. Viral strategies for targeting the central and peripheral nervous systems. Annu. Rev. Neurosci. https://doi.org/10.1146/annurev-neuro-080317-062048 (2018).
Leone, P. et al. Long-term follow-up after gene therapy for canavan disease. Sci. Transl Med. 4, 165ra163 (2012).
Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M. & Abedi, M. R. Gene therapy clinical trials worldwide to 2017: an update. J. Gene Med. 20, e3015 (2018).
Calcedo, R., Vandenberghe, L. H., Gao, G., Lin, J. & Wilson, J. M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 199, 381–390 (2009).
Mingozzi, F. et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422 (2007).
Petry, H. et al. Effect of viral dose on neutralizing antibody response and transgene expression after AAV1 vector re-administration in mice. Gene Ther. 15, 54–60 (2008).
Lu, Y. & Song, S. Distinct immune responses to transgene products from rAAV1 and rAAV8 vectors. Proc. Natl Acad. Sci. USA 106, 17158–17162 (2009).
Ashley, S. N., Somanathan, S., Giles, A. R. & Wilson, J. M. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell Immunol. 346, 103997 (2019).
Mendell, J. R. et al. Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 363, 1429–1437 (2010).
Wilson, J. M. & Flotte, T. R. Moving forward after two deaths in a gene therapy trial of myotubular myopathy. Hum. Gene Ther. 31, 695–696 (2020).
Hinderer, C. et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 29, 285–298 (2018).
Nidetz, N. F. et al. Adeno-associated viral vector-mediated immune responses: understanding barriers to gene delivery. Pharmacol. Ther. 207, 107453 (2020).
Pickar-Oliver, A. & Gersbach, C. A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019).
Mullard, A. First in vivo CRISPR candidate enters the clinic. Nat. Rev. Drug Discov. 18, 656 (2019).
Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S. & Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646 (2010).
Joung, J. K. & Sander, J. D. TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 14, 49–55 (2013).
Willems, J. et al. ORANGE: a CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLoS Biol. 18, e3000665 (2020).
Park, S. H. et al. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 47, 7955–7972 (2019).
Sugaya, K. & Vaidya, M. Stem cell therapies for neurodegenerative diseases. Adv. Exp. Med. Biol. 1056, 61–84 (2018).
Maeder, M. L. et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233 (2019).
Sun, J. et al. CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage. Nat. Commun. 10, 53 (2019).
Popp, M. W. & Maquat, L. E. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell 165, 1319–1322 (2016).
Lau, C. H., Ho, J. W., Lo, P. K. & Tin, C. Targeted transgene activation in the brain tissue by systemic delivery of engineered AAV1 expressing CRISPRa. Mol. Ther. Nucleic Acids 16, 637–649 (2019).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9–cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017).
Levy, J. M. et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 4, 97–110 (2020).
Grunewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019).
Zuo, E. et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat. Methods 17, 600–604 (2020).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Rinaldi, C. & Wood, M. J. A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 14, 9–21 (2018).
Leavitt, B. R. & Tabrizi, S. J. Antisense oligonucleotides for neurodegeneration. Science 367, 1428–1429 (2020).
Chen, G., Katrekar, D. & Mali, P. RNA-guided adenosine deaminases: advances and challenges for therapeutic RNA editing. Biochemistry 58, 1947–1957 (2019).
Abudayyeh, O. O. et al. RNA targeting with CRISPR–Cas13. Nature 550, 280–284 (2017).
Konermann, S. et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676.e14 (2018).
Abudayyeh, O. O. et al. A cytosine deaminase for programmable single-base RNA editing. Science 365, 382–386 (2019).
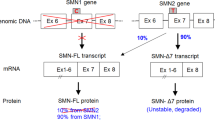
Lefebvre, S. et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165 (1995).
Han, K. J. et al. Ubiquitin-specific protease 9× deubiquitinates and stabilizes the spinal muscular atrophy protein-survival motor neuron. J. Biol. Chem. 287, 43741–43752 (2012).
Passini, M. A. et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl Med. 3, 72ra18 (2011).
Darras, B. T. et al. Nusinersen in later-onset spinal muscular atrophy: long-term results from the phase 1/2 studies. Neurology 92, e2492–e2506 (2019).
Finkel, R. S. et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 377, 1723–1732 (2017).
Finkel, R. S. et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026 (2016).
Naryshkin, N. A. et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345, 688–693 (2014).
Zhou, M. et al. Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific induced pluripotent stem cells. Hum. Gene Ther. 29, 1252–1263 (2018).
Al-Zaidy, S. et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr. Pulmonol. 54, 179–185 (2019).
Sun, J. & Roy, S. The physical approximation of APP and BACE-1: a key event in Alzheimer’s disease pathogenesis. Dev. Neurobiol. 78, 340–347 (2018).
Das, U. et al. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 19, 55–64 (2016).
Huang, Y. A., Zhou, B., Wernig, M. & Sudhof, T. C. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 168, 427–441.e21 (2017).
Doran, E. et al. Down syndrome, partial trisomy 21, and absence of Alzheimer’s disease: the role of APP. J. Alzheimers Dis. 56, 459–470 (2017).
Li, C. & Gotz, J. Tau-based therapies in neurodegeneration: opportunities and challenges. Nat. Rev. Drug Discov. 16, 863–883 (2017).
Tuszynski, M. H. et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11, 551–555 (2005).
Nagahara, A. H. et al. Long-term reversal of cholinergic neuronal decline in aged non-human primates by lentiviral NGF gene delivery. Exp. Neurol. 215, 153–159 (2009).
Rafii, M. S. et al. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol. 75, 834–841 (2018).
Castle, M. J. et al. Postmortem analysis in a clinical trial of AAV2–NGF gene therapy for Alzheimer’s disease identifies a need for improved vector delivery. Hum. Gene Ther. 31, 415–422 (2020).
Nagahara, A. H. et al. MR-guided delivery of AAV2–BDNF into the entorhinal cortex of non-human primates. Gene Ther. 25, 104–114 (2018).
DeVos, S. L. et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl Med. https://doi.org/10.1126/scitranslmed.aag0481 (2017).
Sud, R., Geller, E. T. & Schellenberg, G. D. Antisense-mediated exon skipping decreases tau protein expression: a potential therapy for tauopathies. Mol. Ther. Nucleic Acids 3, e180 (2014).
Velazquez, R. et al. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell https://doi.org/10.1111/acel.12775 (2018).
Zhou, L. et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 8, 15295 (2017).
Yu, J. T., Tan, L. & Hardy, J. Apolipoprotein E in Alzheimer’s disease: an update. Annu. Rev. Neurosci. 37, 79–100 (2014).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Hudry, E. et al. Gene transfer of human APOE isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl Med. 5, 212ra161 (2013).
Rosenberg, J. B. et al. AAVrh.10-mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer’s disease. Hum. Gene Ther. Clin. Dev. 29, 24–47 (2018).
Lin, Y.-T. et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7 (2018).
Gyorgy, B. et al. CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s disease. Mol. Ther. Nucleic Acids 11, 429–440 (2018).
Chang, J. L. et al. Targeting amyloid-β precursor protein, APP, splicing with antisense oligonucleotides reduces toxic amyloid-β production. Mol. Ther. 26, 1539–1551 (2018).
Muller, U. C., Deller, T. & Korte, M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18, 281–298 (2017).
Iyer, S. et al. Precise therapeutic gene correction by a simple nuclease-induced double-stranded break. Nature 568, 561–565 (2019).
Fol, R. et al. Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 131, 247–266 (2016).
Obeso, J. A. et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov. Disord. 23, S548–S559 (2008).
Christine, C. W. et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669 (2009).
Mittermeyer, G. et al. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum. Gene Ther. 23, 377–381 (2012).
San Sebastian, W. et al. Safety and tolerability of magnetic resonance imaging-guided convection-enhanced delivery of AAV2–hAADC with a novel delivery platform in nonhuman primate striatum. Hum. Gene Ther. 23, 210–217 (2012).
Palfi, S. et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146 (2014).
Baron, M. S., Wichmann, T., Ma, D. & DeLong, M. R. Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J. Neurosci. 22, 592–599 (2002).
Emborg, M. E. et al. Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J. Cereb. Blood Flow. Metab. 27, 501–509 (2007).
Luo, J. et al. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science 298, 425–429 (2002).
Kaplitt, M. G. et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet 369, 2097–2105 (2007).
LeWitt, P. A. et al. AAV2–GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 10, 309–319 (2011).
Niethammer, M. et al. Long-term follow-up of a randomized AAV2–GAD gene therapy trial for Parkinson’s disease. JCI Insight 2, e90133 (2017).
Collier, T. J. & Sortwell, C. E. Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging 14, 261–287 (1999).
Chen, Y. H. et al. MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J. 22, 261–275 (2008).
Su, X. et al. Safety evaluation of AAV2–GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum. Gene Ther. 20, 1627–1640 (2009).
Marks, W. J. Jr. et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 7, 400–408 (2008).
Marks, W. J. Jr et al. Gene delivery of AAV2–neurturin for Parkinson’s disease: a double-blind, randomised, controlled trial. Lancet Neurol. 9, 1164–1172 (2010).
Bartus, R. T. et al. Bioactivity of AAV2–neurturin gene therapy (CERE-120): differences between Parkinson’s disease and nonhuman primate brains. Mov. Disord. 26, 27–36 (2011).
Bartus, R. T. et al. Safety/feasibility of targeting the substantia nigra with AAV2–neurturin in Parkinson patients. Neurology 80, 1698–1701 (2013).
Decressac, M. et al. α-synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl Med. 4, 163ra156 (2012).
Qian, H. et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556 (2020).
Wong, Y. C. & Krainc, D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13 (2017).
Soldner, F. et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99 (2016).
Fuchs, J. et al. Genetic variability in the SNCA gene influences α-synuclein levels in the blood and brain. FASEB J. 22, 1327–1334 (2008).
Zharikov, A. D. et al. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 125, 2721–2735 (2015).
Uehara, T. et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting α-synuclein as a novel therapy for Parkinson’s disease. Sci. Rep. 9, 7567 (2019).
Kantor, B. et al. Downregulation of SNCA expression by targeted editing of DNA methylation: a potential strategy for precision therapy in PD. Mol. Ther. 26, 2638–2649 (2018).
Benskey, M. J. et al. Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front. Mol. Neurosci. 11, 36 (2018).
Gorbatyuk, O. S. et al. In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 18, 1450–1457 (2010).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Do, J., McKinney, C., Sharma, P. & Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 14, 36 (2019).
Sardi, S. P. et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl Acad. Sci. USA 110, 3537–3542 (2013).
Morabito, G. et al. AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol. Ther. 25, 2727–2742 (2017).
Gandhi, P. N., Chen, S. G. & Wilson-Delfosse, A. L. Leucine-rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 87, 1283–1295 (2009).
Hatcher, J. M., Choi, H. G., Alessi, D. R. & Gray, N. S. Small-molecule inhibitors of LRRK2. Adv. Neurobiol. 14, 241–264 (2017).
Ness, D. et al. Leucine-rich repeat kinase 2 (LRRK2)-deficient rats exhibit renal tubule injury and perturbations in metabolic and immunological homeostasis. PLoS ONE 8, e66164 (2013).
Fuji, R. N. et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl Med. 7, 273ra215 (2015).
Baptista, M. A. et al. Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE 8, e80705 (2013).
Zhao, H. T. et al. LRRK2 antisense oligonucleotides ameliorate α-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol. Ther. Nucleic Acids 8, 508–519 (2017).
Ross, C. A. & Tabrizi, S. J. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 10, 83–98 (2011).
Kordasiewicz, H. B. et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044 (2012).
Tabrizi, S. J. et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 380, 2307–2316 (2019).
Rodrigues, F. B., Ferreira, J. J. & Wild, E. J. Huntington’s disease clinical trials corner: June 2019. J. Huntingtons Dis. 8, 363–371 (2019).
Evers, M. M. et al. AAV5–miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol. Ther. 26, 2163–2177 (2018).
Boudreau, R. L. et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 17, 1053–1063 (2009).
Stanek, L. M. et al. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 25, 461–474 (2014).
McBride, J. L. et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 19, 2152–2162 (2011).
Grondin, R. et al. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 135, 1197–1209 (2012).
Wang, G., Liu, X., Gaertig, M. A., Li, S. & Li, X. J. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc. Natl Acad. Sci. USA 113, 3359–3364 (2016).
Dietrich, P., Johnson, I. M., Alli, S. & Dragatsis, I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 13, e1006846 (2017).
Shin, J. W. et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 25, 4566–4576 (2016).
van Bilsen, P. H. et al. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum. Gene Ther. 19, 710–719 (2008).
Southwell, A. L. et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol. Ther. 22, 2093–2106 (2014).
Pfister, E. L. et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr. Biol. 19, 774–778 (2009).
Rodrigues, F. B. & Wild, E. J. Huntington’s disease clinical trials corner: February 2018. J. Huntingtons Dis. 7, 89–98 (2018).
Zeitler, B. et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142 (2019).
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R. & Gitler, A. D. ALS genetics: gains, losses, and implications for future therapies. Neuron https://doi.org/10.1016/j.neuron.2020.08.022 (2020).
Bruijn, L. I. et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854 (1998).
Smith, R. A. et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 116, 2290–2296 (2006).
Miller, T. M. et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013).
Miller, T. et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119 (2020).
Foust, K. D. et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. 21, 2148–2159 (2013).
Gaj, T. et al. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 3, eaar3952 (2017).
Mueller, C. et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N. Engl. J. Med. 383, 151–158 (2020).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Haeusler, A. R., Donnelly, C. J. & Rothstein, J. D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395 (2016).
Jiang, J. et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550 (2016).
Tuszynski, M. H. et al. Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147 (2015).
Christine, C. W. et al. Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann. Neurol. 85, 704–714 (2019).
McFarthing, K., Prakash, N. & Simuni, T. Clinical trial highlights: 1. Gene therapy for Parkinson’s, 2. Phase 3 study in focus—INTEC Pharma’s accordion pill, 3. Clinical trials resources. J. Parkinsons Dis. 9, 251–264 (2019).
Darras, B. T. et al. An integrated safety analysis of infants and children with symptomatic spinal muscular atrophy (SMA) treated with nusinersen in seven clinical trials. CNS Drugs 33, 919–932 (2019).
Mercuri, E. et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 378, 625–635 (2018).
Acknowledgements
The authors thank L. Parra, B. Aulston, D. Galasko, and J. Brewer (all at UCSD) for comments on the manuscript. The authors apologize that due to space restrictions, primary papers could not be cited in most cases. Work in the Sun Lab is supported by NSFC grants 82071193 and 32000673. Work in the Roy Lab is supported by grants from the NIH (R01AG048218, R01NS11978, R01NS075233, R21AG052404 and UG3NS111688) and the US–Israel Binational Science Foundation (BSF, number 2019248).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
J.S. and S.R. have applied for patents related to gene editing in AD (US patent application number 16251970). S.R. is also the scientific founder of, advisor to, and owns equity in CRISPRAlz.
Additional information
Peer review information Nature Neuroscience thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sun, J., Roy, S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci 24, 297–311 (2021). https://doi.org/10.1038/s41593-020-00778-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-020-00778-1
This article is cited by
-
Role of the P2 × 7 receptor in neurodegenerative diseases and its pharmacological properties
Cell & Bioscience (2023)
-
Current Update on Categorization of Migraine Subtypes on the Basis of Genetic Variation: a Systematic Review
Molecular Neurobiology (2023)
-
The Role of Oxidative Stress in Trisomy 21 Phenotype
Cellular and Molecular Neurobiology (2023)
-
Non-viral approaches for gene therapy and therapeutic genome editing across the blood–brain barrier
Med-X (2023)
-
Proteinopathies associated to repeat expansion disorders
Journal of Neural Transmission (2022)
