
Abstract
The early stages of the virus–cell interaction have long evaded observation by existing microscopy methods due to the rapid diffusion of virions in the extracellular space and the large three-dimensional cellular structures involved. Here we present an active-feedback single-particle tracking method with simultaneous volumetric imaging of the live cell environment called 3D-TrIm to address this knowledge gap. 3D-TrIm captures the extracellular phase of the infectious cycle in what we believe is unprecedented detail. We report what are, to our knowledge, previously unobserved phenomena in the early stages of the virus–cell interaction, including skimming contact events at the millisecond timescale, orders of magnitude change in diffusion coefficient upon binding and cylindrical and linear diffusion modes along cellular protrusions. Finally, we demonstrate how this method can move single-particle tracking from simple monolayer culture toward more tissue-like conditions by tracking single virions in tightly packed epithelial cells. This multiresolution method presents opportunities for capturing fast, three-dimensional processes in biological systems.
Similar content being viewed by others

Main
The ongoing SARS-CoV-2 pandemic has demonstrated with frightening clarity the need for fundamental research in physical virology to exploit and counter the mechanisms of viral infection. The first point of contact with the host organism occurs in the extracellular space of the epithelia, whose cells form a tightly packed arrangement protected by an extended mucus layer and glycocalyx1,2,3,4,5. The structure of the extracellular matrix (ECM) has been shown to be critical to the viral lifecycle, undergoing changes in structure and composition upon introduction of viral pathogens6 and hosting biofilm-like viral assemblies for cell-to-cell transmission7.
Single-particle tracking (SPT) methods have emerged as a powerful tool in our understanding of viral infection8,9,10,11,12. These methods have uncovered virion binding mechanisms to the cell surface13,14, distinguished internalization pathways15,16,17,18,19, identified the cellular location of envelope fusion20,21,22,23,24,25,26, characterized cytoskeletal trafficking14,19,20,21,22,27,28,29,30,31,32,33 and demonstrated how viruses hijack filopodia to efficiently infect neighboring cells19,23,34,35,36,37,38,39.
Despite these advances, it has thus far not been possible to follow virions in the extracellular space with sufficient detail to interrogate this important phase of viral infection. This is because extracellular diffusion occurs across depth ranges exceeding 10 μm and at diffusive speeds 2–3 orders of magnitude greater than the highly confined processes that can be followed by conventional SPT methods. Even the most advanced image-based SPT methods such as spinning-disk confocal (SDC) and light-sheet microscopy40,41 are unable to meet these challenges as they suffer from limitations caused by attempting to simultaneously track and image disparately scaled objects on a single platform.
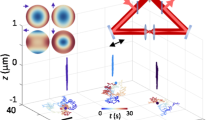
One way to understand these limitations is to consider the sampling of trajectory points in terms of the number of localizations per second (loc s−1), which for image-based tracking methods is given by the volumetric imaging rate (Fig. 1a). As volumetric data are acquired frame by frame, the temporal resolution scales poorly as the axial extent of the process in question grows. Yet shrinking the volume Z size reduces the likelihood that the virion will remain in the volumetric field of view, shortening the observation duration and, thus, the absolute number of localizations acquired. Additionally, as camera exposure times decrease, photon limitations when trying to image single particles become a limiting factor. Given a modest value of 16 z-slices spaced over an axial range sufficient to capture extracellular diffusion, even the fastest light-sheet systems can theoretically acquire as many as 6.25 loc s−1 at best. Overcoming these fundamental limitations in speed and axial range motivated the development of active-feedback tracking methods which focus exclusively on the tracked particle42. Such methods can localize particles with high spatial and temporal resolution, but the cost to obtaining this enhanced temporal resolution is loss of environmental context. While such methods have been useful in studying particle dynamics in isolation, this missing context has precluded the application of such methods to studying particle–environment interactions.

a, Experimental setup. Fluorescently labeled VLPs are added to live cells plated on a coverslip. The sample is placed on a heated sample holder mounted on a piezoelectric stage. Inset, sampling rate comparison among spinning disk, light sheet and 3D-TrIm. FPS, frames per second. b, Overview of 3D-SMART tracking of single viruses. The EOD and TAG lens rapidly scan the focused laser spot around the local particle area. Photon arrival times and the current laser position are used to calculate the position of the virus within the scan area. Using the measured position, the piezoelectric stage moves to recenter the virus within the scan area. c, Concept of 3D-FASTR volumetric imaging. By outfitting a traditional two-photon LSM with an ETL, a repeatable, tessellated 3D sampling pattern can be generated during each frame-time. Over a set number of frame-times, the entire volume is sampled. d, Construction of global volumes in 3D-TrIm. As the virus diffuses, 3D-SMART moves the sample, and the 3D-FASTR imaging system collects sequential volumes from different areas around the particle (black dot). These time-resolved local volumes can be used to generate an integrated global volume (see also Supplementary Video 1).
To address this gap, we present 3D Tracking and Imaging Microscopy (3D-TrIm), a multi-modal instrument that integrates a real-time active-feedback tracking microscope43,44,45 with a volumetric imaging system46 capable of simultaneously tracking the high-speed dynamics of extracellular virions while imaging the surrounding three-dimensional (3D), live cell environment (Fig. 1a). This multi-modal approach provides unique value which cannot be extracted by its independent components alone and, as demonstrated here, provides unprecedented glimpses into the initial moments of the virus–cell interaction. Using 3D-TrIm, we acquired over 3,000 viral trajectories and demonstrate the benefits of how this contextualized tracking uses localizations in time analogously to super-resolution methods. This ‘super-temporal resolution’ enables not just the formation of trajectories, but the detection of changes in diffusive regimes and the tracing out of nanoscale structural features. Finally, 3D-TrIm was applied to advance SPT from monolayer cell culture to tightly packed, 3D epithelial culture models, providing a window into this critical stage of viral infection.
Results
Capturing extracellular viral dynamics requires the ability to track a rapidly diffusing target in three dimensions. To accomplish this, we apply the recently developed 3D single-molecule active real-time tracking (3D-SMART)43,44. 3D-SMART is an active-feedback microscopy method that uses real-time position information to ‘lock on’ to moving fluorescent targets. Critically, 3D-SMART can capture particles diffusing at up to 10 μm2 s−1 with only a single fluorophore label43, making it an ideal choice for capturing diffusing virions45.
The implementation of 3D-SMART in the current work is similar to previous reports. Briefly, a rapid 3D laser scan is generated by an electro-optic deflector (EOD) and tunable acoustic gradient (TAG) lens. The scanned laser spot excites photons from the fluorescently labeled diffusing viral particle (Fig. 1b and Supplementary Video 1). Photons are collected on a single-photon-counting avalanche photodiode (APD), which also acts as a pinhole to provide confocal detection and prevent detection of out-of-focus virions. The photon arrival times are used to calculate the real-time position every 20 µs which is then used in an integral feedback loop to move the sample via a piezoelectric stage, effectively fixing the moving target in the focus of the microscope objective. In the event that a second virion diffuses into the tracking volume, a spike in intensity is recorded. Trajectories are split at these ‘jump’ points so that each trajectory represents a single virion. 3D-SMART produces a 3D particle trajectory with 1,000 loc s−1, limited here by the 1-ms mechanical response time of the stage, with localization precision up to ~20 nm in XY and ~80 nm in Z (Supplementary Fig. 1) and duration only limited by the travel range of the piezoelectric stage (75 × 75 × 50 µm3; XYZ).
While 3D-SMART is ideally suited for capturing the fast dynamics of extracellular viral particles over vast axial ranges, alone it lacks environmental context. A complementary rapid volumetric imaging method is needed to capture the live cell environment. The choice of imaging system is nontrivial as the active-feedback nature of 3D-SMART requires a moving stage which places two constraints that must be considered. First, imaging with camera-based methods, such as light-sheet microscopy, would result in motion blur across a single exposure time; the chosen imaging modality must be faster than the 1-ms response time of the piezoelectric stage. Second, the stage cannot be stepped to perform ‘z-stacking’ for volumetric imaging.
To meet these criteria, we implement 3D Fast Acquisition by z-Translating Raster (3D-FASTR) microscopy to provide rapid volumetric imaging around the tracked viral particle46. 3D-FASTR uses a two-photon laser scanning microscope outfitted with an electrically tunable lens (ETL). The short pixel dwell time of the raster scan (~1 µs) prevents motion blur, and the ETL performs remote focusing of the imaging laser for 3D imaging. In contrast to a conventional z-stack, 3D-FASTR performs a continuous focal scan across an 8-µm range. At optimized scan frequencies, the combination of the XY raster scan and the Z focal scan evenly samples voxels in a tessellating pattern which tiles to scan the entire volume (Fig. 1c and Supplementary Video 1). At short acquisition times, unsampled voxels will have many sampled neighbors such that the volumetric imaging rate can be increased 2–4-fold over conventional image stacking methods through interpolation46. The ability to rapidly image volumes without moving the sample makes 3D-FASTR the perfect complement to 3D-SMART.
To integrate the two microscopes into a combined platform, excitation and emission for both tracking and imaging are routed through a single shared objective lens (OL, ×100, numerical aperture = 1.4) on an inverted microscope stand outfitted with a 3D piezoelectric stage. The piezoelectric stage is mounted on a secondary motorized stage which remains static during acquisition but can be moved between trajectories to capture different areas of the sample (Extended Data Fig. 1). Registration of the 3D-SMART and 3D-FASTR systems is required to generate combined 3D-TrIm datasets (Supplementary Figs. 2 and 3). The piezoelectric stage represents a shared spatial grid between the two microscopes; the real-time stage position is a measure of the tracked particle’s location which is tagged to the imaging XYZ voxel position. The motion of the piezoelectric stage translates the 3D imaging field of view, expanding the total imaging volume size based on how far the virion diffuses. This effect is most dramatic in Z where the 8-µm 3D-FASTR imaging range moves across the 50-µm Z stage range. These tagged positions are then used with calibration information (Supplementary Figs. 4 and 5) to place each image voxel in the shared global volume space. This per-voxel registration allows for an arbitrary volume acquisition rate; voxels can be accumulated over the duration of an entire trajectory to form a single global volume or spread across multiple local volumes over time (shown here at ~17 s per volume) to construct sequences over the course of the trajectory (Fig. 1d).
For this pioneering study into the early stages of the virus–cell interaction, we investigated the journey of vesicular stomatitis virus (VSV)-G-pseudotyped lentiviruses from the extracellular solution to the cell surface and beyond. This specific virus-like particle (VLP) was selected for its wide cellular tropism due to the ubiquitous host entry partner of VSV-G, the low-density lipoprotein receptor (LDLR)47, which has established VSV-G as the standard for gene delivery48. We utilized an efficient and well-characterized internal virus labeling approach whereby fluorescent proteins fused to the HIV-1 viral protein R (Vpr) are packaged into budding lentiviruses49. Successful incorporation of eGFP-Vpr into single virions was confirmed by tracking VSV-G eGFP-Vpr in the absence of cells and by immunofluorescence experiments (Supplementary Figs. 6–8). Based on a photobleaching analysis that indicates a single eGFP-Vpr has an intensity of ~2 kHz, each VLP tracked in this study has somewhere between 10 and 100 eGFP-Vpr packaged within its capsid (Supplementary Fig. 9). Critically, these VLPs were shown to remain infective (Supplementary Fig. 10).
To initiate data collection, the 3D-TrIm microscope locates diffusive virions by searching a plane approximately 5 μm above the apical cell surface so that viruses are captured before virus–cell first contacts. When a VLP enters the 3D-SMART tracking volume, a large increase in detected photons above the background level is observed on the single-photon counter, which triggers the active-feedback tracking loop and 3D-FASTR image acquisition.
Super-temporal resolution of viral trajectories in context
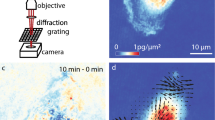
To demonstrate the impact of this method, extracellular trajectories of rapidly diffusing extracellular VLPs using both 3D-TrIm and an Andor Dragonfly SDC microscope were acquired. The side-by-side data shown in Fig. 2a,b demonstrate the vast difference in the amount of data available from 3D-TrIm compared with SDC data (see also Extended Data Fig. 2). The SDC microscope shows the trajectories of multiple extracellular virions, the majority of which remain in the trackable field of view long enough to obtain only a few sample points. Such sample points are characterized as long, straight lines averaging the behavior between frame exposures. Tracking and imaging are coupled together so the field of view is confined to a fixed area and only one localization can be made per volume acquired. The overlaid volumes show that over three acquired volumes there are three localizations. In contrast, the 3D-TrIm trajectory of a single virion features thousands of sample points, capturing the contour of motion instead of straight lines (Fig. 2b). This localization rate and field of view are completely decoupled from the volume rate. Instead, over the course of three volumes, there are thousands of localizations per volume and the imaging field of view moves with the particle, reducing bleaching. Notably, the utility of this super-resolved tracking is not just in the ability to form trajectories but also in the ability to extract quantitative and qualitative meaning from the data. This super-temporally resolved data can extract not just a single approximated diffusion coefficient, but can detect changes in diffusion speed and mode due to interactions.

a, Extracellular viral diffusion collected on an Andor Dragon fly SDC microscope. The same area was sampled continuously. Each volume has a depth of 8 μm split into 16 z planes. Virus particles (eGFP-Vpr VSV-G) and live cells (HeLa, SiR650-Actin stain) were imaged simultaneously with a camera exposure time of 40 ms. b, Extracellular viral diffusion collected on 3D-TrIm microscope. A single virus particle (eGFP-Vpr VSV-G) was tracked continuously (1 ms of sampling shown) and the surrounding area was imaged 5 μm below the particle and 3 μm above, to give an approximate 8-μm volume of cellular environment (HeLa, SiR650-Actin stain). The resulting trajectories over the entire acquisition period are shown and color-coded by time. c,d, 3D reconstruction of a single VSV-G VLP trajectory with live HeLa cells stained with SYTO61 from a four-dimensional (4D) dataset. Cells are color-coded by intensity and distance from the cell surface to the virus trajectory in c and d, respectively. Circular inset, enlarged view of ‘skimming’ event. e, Correlation between diffusivity and cell-to-virus distance. Skimming events are highlighted in purple. Skimming event shown in circular inset of d occurs at 68–78 s. f, Top-down (xy) complete imaging area with trajectory. g, Lateral (yz) view with trajectory color-coded by diffusion coefficient segments calculated by change-point analysis. h,i, Identical to c and d except VSV-G trajectory coregistered with live GM701 fibroblast cells stained with SiR-Actin (see also Supplementary Video 2). Circular inset, enlarged view of ‘skimming’ event. j, Lateral (yz) view of the trajectory shown in h, demonstrating that the VLP is tracked over a depth of 30 μm.
Extracellular virions skim the live cell surface
An example trajectory of these early interactions is shown in Fig. 2c,f,g, which features live HeLa cells fluorescently labeled with the nucleic acid stain SYTO61. Taking advantage of the coregistered individual local volumes (Extended Data Fig. 3), a global 3D render of the cells was constructed using the maximum intensity of each voxel over time. The viral trajectory is constructed from the piezoelectric stage coordinates and visualized here at the 3-ms sampling intervals used for diffusivity calculations. The single VSV-G VLP was initially captured in the search plane and diffused for ~90 s, reaching a maximum height of nearly 20 μm above the cell surface. The virion closely approaches and intermittently touches the surface of several cells over the course of the trajectory.
These periods of transient contact (‘skimming’) were quantified in two ways. First, change-point analysis was applied to identify changes in the diffusive state of the VLP as it traverses the volume50,51. The extracted diffusion coefficients are shown as color-coded lines in Fig. 2e. Second, the distance between the VLP and the cell surface was calculated using segmented volume data (Extended Data Fig. 4). This VLP–cell distance is visualized in Fig. 2d, a color-mapped volume with yellow/red indicating approach within 1 μm/0.5 μm (comparable to the axial extent of the 3D-FASTR point-spread function, Supplementary Fig. 11), respectively. This value of 0.5 μm was used as the threshold value for determining contact. The trajectory reveals multiple and repeated contact events at various surface locations (highlighted in purple in Fig. 2e). While the HeLa cell shown is LDLR-positive, these skimming events are not restricted to receptor-positive cells but are also observed in GM701 fibroblasts (low LDLR expression level) (Fig. 2h–j and Supplementary Video 2). Additional examples of viral skimming can be found in Extended Data Fig. 5 and Supplementary Figs. 12–15.
These transient contacts were quantified to yield their occurrence (Supplementary Fig. 16a), frequency (Supplementary Fig. 16b) and dwell time (Supplementary Fig. 17) for three different cell types of varying LDLR expression level and morphology. These contacts can be as short as several milliseconds in duration but numerous per trajectory, without promoting long-duration viral attachment. Exponential fitting of contact event durations on HeLa cells yielded two populations with dwell times of 23 ± 1 and 102 ± 5 ms (Supplementary Fig. 17a). The shorter dwell time is consistent with free diffusion, while the longer dwell time indicates an interaction of the VLP with the cell surface or ECM. These dwell times were longer for HeLa (102 ± 5 ms) compared with the low-LDLR GM701 fibroblast cells (88 ± 8 ms, Supplementary Fig. 17b), though they exhibited identical fast components. Since these two diffusive states are not correlated with the presence (BJ) or absence (GM701) of LDLR, it can be concluded that these initial interactions are not related to receptor priming but potentially related to cell morphology (Supplementary Fig. 17c).
The increased dwell time near the cell surface also correlated with slower diffusion in the presence of HeLa cells. A trajectory-wide analysis of ‘skimming’ VSV-G VLPs showed 80% of trajectories acquired in the presence of HeLa cells have diffusion coefficients within the range of 0.5–3.4 µm2 s−1 (Supplementary Fig. 18a), consistent with diffusive rates in the presence of cells in previous reports13,14,51. Interestingly, VLPs exhibited increased diffusivity of 1.3–4.5 µm2 s−1 for thinner fibroblast cells, revealing a cell-type dependence on extracellular diffusivity (Supplementary Fig. 18a, two-sample t-test, P < 0.01). The unique correlative imaging and high-speed tracking capabilities of the 3D-TrIm microscope enabled a quantitative examination of the relationship between VLP diffusion and distance from the cell surface. This correlation analysis revealed a distance-dependent relationship, with VLPs slowing down as they near the cell surface. Statistically significant inhibition of diffusion was observed within 2.5 µm of the cell surface for all cell types tested (Supplementary Fig. 18e). Again, compared with the fibroblasts, VLPs in the presence of HeLa trended toward lower diffusion coefficients near the cell surface (Supplementary Fig. 18f). This difference again suggests morphological effects imposed by the more irregularly shaped HeLa compared with flatter, more uniform fibroblasts.
While all three cell types showed a trend toward slower diffusion near the cell surface, each still displayed a substantial fraction of VLPs with segments of diffusion >1 µm2 s−1 at less than 1 µm from the cell surface. Again, this effect was cell-type dependent, with BJ cells showing a much higher percentage of fast-diffusing VLPs near the surface (BJ: 33.6% ± 11.8%, HeLa: 16.8% ± 3.9%, GM701: 16.7% ± 7.3%). This fast diffusion near the cell surface suggests a complex interplay between the extracellular environment and VLP dynamics. Such short-lived contact events are resolvable with 3D-TrIm due to the unique combination of super-temporally resolved tracking combined with 3D Imaging which can discern cellular contact on the millisecond timescale.
3D-TrIm captures the transitional dynamics of binding
In addition to the transient contacts observed above, long-term binding events of individual VSV-G VLPs on the cell surface were also captured by 3D-TrIm. Unlike skimming, these binding events display dramatic diffusivity changes upon contact. In the first example (Fig. 3a–c, Supplementary Fig. 19 and Supplementary Video 3), the virus initially undergoes several skimming events, marked by close approach to the surface of multiple cells with subtle changes in the diffusion coefficient. At ~70 s, as the virus–cell distance reaches a minimum, viral diffusivity drops by two orders of magnitude and remains bound for several minutes (indicated by the pink box in Fig. 3d). In such cases where motion of the particle is confined, the observation duration is limited by photobleaching of the virion to several minutes at the reported laser power. Photobleaching of the cells occurs over a longer duration due to sampling a larger area less frequently.

a–c, 3D-TrIm trajectory of a single VSV-G VLP binding to live nucleic acid-labeled HeLa cells. a, Intensity volume render (see also Supplementary Video 3). Gray circular inset shows moment of VLP binding to the cell surface. b, Virus–cell distance render. c, Maximum intensity projection (MIP) with trajectory overlay. d, Trajectory diffusivity and virus–cell distance–time trace. Skimming events are highlighted in purple; binding events are highlighted in pink. e–g, 3D-TrIm trajectory of VSV-G VLP transient binding to live actin-labeled GM701 cells. e, Intensity volume render with trajectory color-coded by diffusivity (see also Supplementary Video 4). f, Close-in view of VLP binding sites. g, MIP with trajectory overlay.
It was also observed that bound viral particles detach after landing and diffuse away or even bind again elsewhere. Figure 3e–g shows a VLP initially bound between two protrusions on an actin-stained GM701 fibroblast (see also Supplementary Fig. 20 and Supplementary Video 4). This bound state (D ≈ 0.04 µm2 s−1; Fig. 3e,f, yellow) persists for ~23 s, before detaching to free diffusion (D > 2 μm2 s−1). After only ~4 s of free diffusion, viral diffusivity drops to less than 0.1 µm2 s−1 upon contact and remains bound for several seconds before detaching again to free diffusion and ultimately leaving the trackable volume (additional example: Extended Data Fig. 6). Combined with the millisecond-scale dwell times above, these multiple and long-term binding events suggest that cell–VLP attachment events cover a large range of timescales.
3D-TrIm traces out nanoscale cellular protrusions
In addition to the ability of 3D-TrIm to resolve milliseconds-long contact and sample changes in diffusive regime, 3D-TrIm’s highly sampled trajectories can trace out nanoscale structures on the cell surface. Similar to how super-localization of emitters enables nanoscale resolution of cellular structures in super-resolution methods, the high 3D precision and rapid sampling of 3D-SMART turns the virus into a nanoscale pen that draws out local features smaller than the diffraction limit which can be contextualized through the simultaneous larger-scale imaging.
One example is the interaction between VLPs and cylindrical protrusions on the cell membrane. Using 3D-TrIm, viral particles were observed binding directly to these structures from the extracellular space (Fig. 4a–e, Extended Data Fig. 7, Supplementary Fig. 21 and Supplementary Video 5). While not stained and therefore not visible in the image, the virus’s path along the surface creates a high-resolution map, carving out the nanoscale cylindrical morphology of the protrusion surface (Fig. 4d,e). The feature traced by this VLP protrudes ~1 μm vertically from the cell surface (Fig. 4d). A best-fit cylinder gives a radius of 105 ± 8.4 nm (Fig. 4e), consistent with the observed size of filopodial protrusions, and, notably, demonstrating the ability for 3D-TrIm to super-resolve tracked features beyond the diffraction limit. These structures were dynamic and resulted in changing cylindrical structure within the viral trajectory over time (Fig. 4f–i), with radii tapering for more distal parts of the structure. VLPs on these structures were consistently measured to have diffusivity values ~0.01 μm2 s−1 (Extended Data Fig. 8 and Supplementary Figs. 22 and 23), nearly identical to other types of membrane diffusion observed (Supplementary Fig. 24). Viral trajectories collected with 3D-TrIm were also able to super-resolve hemispherical membrane blebs (Fig. 4j,k and Supplementary Fig. 25), highlighting again how the high-sampling-rate 3D tracking can result in super-resolution structural tracing.

a, VSV-G landing on protrusion from the surface of a live SYTO61-stained HeLa cell (see also Supplementary Video 5). b, Magnified view with slices through x and y axes. c, Top-down (xy) complete imaging area with trajectory. d, Magnified views showing the hollow cylinder the VLP draws out while diffusing around the protrusion. e, Cylindrical fitting of the bound portion of the trajectory in a–d (16–133 s), radius = 105 ± 8.4 nm. f, Additional high-resolution trajectory of VLP on a protrusion captured after landing. g, Lateral (yz) view. VLP tracking captures change in protrusion shape. h, Enlarged views showing VLP progression after starting from a proximal position on the membrane. i, Cylindrical fitting of base (10–31 s) and extremity (55–99 s) of the trajectory in f–h, radius = 153 ± 8.7 nm and 77 ± 8.4 nm, respectively. Average protrusion radius of 125 ± 16 nm (n = 5). j, Top-down view of VSV-G VLP diffusing on membrane bleb of actin-stained BJ fibroblast cells. Inset: enlarged view. k, 3D volume rendering for data shown in j, except trajectory colored by diffusion coefficient.
VLP dynamics in the epithelia: beyond monolayer cell culture
As is typical of SPT experiments, the previous examples were captured in the presence of monolayer cultures, which have been shown to differ from more realistic tissue models52. Here, we demonstrate the potential for 3D-TrIm to operate in complex environments at considerable depths in systems that more closely approximate the infection routes of viruses in vivo. A system of particular relevance to the extracellular dynamics of viruses (and to respiratory viruses in particular) is the epithelium, which is protected by a thick mucus layer1,2 and a size-excluding periciliary layer53. For well-differentiated epithelial cells to form tightly packed arrangements in vitro, they must be grown on a semipermeable membrane support to allow access to the basolateral layer and provide a more realistic growth environment. The thick (>10 µm), tightly packed epithelial layer cannot be grown directly on a coverslip, making observation of dynamics in these critical systems impossible in conventional microscopy methods. In contrast, 3D-TrIm’s large axial range enables unprecedented high-speed SPT in these more biologically relevant tissue models.
Epithelial model systems were prepared by growing HT29-MTX cells on a semipermeable membrane support and inverted so that the cells were suspended above the coverslip surface (Supplementary Fig. 26). The diffusion of viruses into this tightly packed layer can be captured from this vantage with millisecond temporal resolution (Fig. 5a–d, Extended Data Fig. 9, Supplementary Fig. 27 and Supplementary Video 6). The VLP experiences confinement near the surface of multiple cells for nearly 3 min. However, it is important to note the difference between subdiffusion/confinement and diffusivity. The average diffusion coefficient for VSV-G traveling in this complex environment is 1.29 ± 0.44 µm2 s−1, in the range of freely diffusing virus particles (Supplementary Fig. 8g) and at least two orders of magnitude higher than bound or internalized processes, suggesting the ECM does not present a substantial physical barrier. Critically, despite this confinement, the virion manages to diffuse more than 10 µm across the Z axis during the trajectory.

a, Top, 3D reconstruction from a 4D dataset covering 10 local volumes, at 10 frames per volume (see also Supplementary Video 6), of suspended HT29-MTX cells grown on inverted matrix support and labeled with SYTO61. Coregistered VSV-G VLP trajectory color-coded by time. Bottom, lateral (xz) view. b, Top-down (xy) MIP. c, Right, magnified cut-through view showing VSV-G VLP confined within vacancy as it diffuses around the edge of tightly packed epithelial cells. Left, isosurface render of cells highlighting sample density. d, Top, the distance of the trajectory to the cell surface is projected as a distance map on the cell volume, with the trajectory color-coded by time overlayed. Bottom, volume with trajectory omitted, showing areas of close contact on the cell surface. e, Left, the resulting trajectory is color-coded by diffusion coefficient, using mean squared displacement and change-point analysis. Right, trajectory with volume omitted, showing highly sampled trajectory across a large range in diffusivity. f, The same fluorescently labeled VLPs (eGFP-Vpr VSV-G) were tracked in 40% glycerol (v/v) on an Andor Dragon fly SDC microscope, with a camera exposure time of 40 ms. A single volume (~8 μm split into 16 z planes) was sampled continuously. The resulting trajectories over the entire acquisition period are shown and color-coded by time.
This large accessible depth range coupled with near-free diffusivity precludes study with even advanced image-based tracking. To demonstrate the benefit of 3D-TrIm in studying this diffusive regime, VLPs were suspended in 40% glycerol, which based on solution-phase tracking should reduce the particle diffusion to ~0.75 µm2 s−1. Figure 5f shows that when imaged at this similar diffusivity, the slower volume sampling of the spinning disk makes it impossible to extract details of local diffusion from the undersampled trajectory. In contrast, the highly sampled trajectory from 3D-TrIm reveals significant (and previously invisible) changes in virion dynamics as it hugs the cell surface (Fig. 5e).
This rapid but trapped diffusion is in stark contrast to viruses near monolayer cultured cells, where the rapid diffusion limits the dwell time near the cell surface. Notably, this example demonstrates the capability for 3D-TrIm to track at low signal levels (35 kHz) in complex environments, which is a notable advantage over other previous active-feedback tracking methods, which require several hundred kHz for successful tracking51,54.
We note here that there are several features of 3D-TrIm that make it extendable to future studies. First, not restricted to capturing rapid extracellular dynamics, 3D-TrIm’s high-resolution tracking ability extends to the later, internalized stages of the infectious cycle (Fig. 6, Supplementary Fig. 28 and Supplementary Video 7). These data demonstrate that the VLPs in this study exhibit the hallmarks of infectious virions in viable cells, undergoing normal cellular processes such as endocytosis and intracellular trafficking. 3D-TrIm is fully compatible with live cell studies and active biological processes.

a, Whole-volume visualization of HeLa cells stained with SiR650 labeling F-actin. Two slice planes were used to section the YZ plane and create a channel that renders the interior volume section semitransparent, revealing the trajectory embedded beneath the cell surface. b, Close-in view of VLP trajectory. c, Top-down XY view of internalized viral trajectory shows high-intensity, actin-rich regions which the trajectory moves around. d, YZ slice view demonstrates that VLP is truly internalized inside the depths of the cell (see also Supplementary Video 7).
Second, 3D-TrIm can observe single-particle dynamics within the same area over long periods to expand beyond the single-particle nature intrinsic to active-feedback tracking methods42. Such prolonged live cell imaging is made possible by a combination of continual laser scanning and viral motion, which reduces the overall laser dwell time at any single position. Multiple-trajectory registration enables 3D-TrIm to accumulate a population of trajectories sufficient to perform statistical analyses on the dynamics and activity of virions in a single area (Extended Data Fig. 10, Supplementary Fig. 29 and Supplementary Video 8). Extended Data Fig. 10b shows the population average (27 of 38 trajectories) of VLP position and diffusivity, revealing that different VLPs frequent similar areas of the cell surface and ECM. Further experiments may uncover what factors influence the heterogenous dwell time of virions to these particular regions of the surface.
Discussion
The data collected here by the 3D-TrIm microscope represent a dramatic step forward for SPT at high speeds in complex systems. Before this study, the immobilization and detachment behaviors of single virions could be observed by epifluorescence microscopy13,55. However, here 3D-TrIm gives a continuous and uninterrupted timeline of the viral particle through the entire process, complete with volumetric imaging and environmentally contextualized diffusion analysis. While in the current study 3D-TrIm is able to track viral particles for minutes at a time, following one particle through the entire infection process will require 10 s of minutes of observation time. This limits the ability of 3D-TrIm to correlate these newly uncovered extracellular and cell-surface dynamics with the infectivity of a single VLP. To address this in the future, we have recently developed an information-efficient sampling approach which extends the observation time dramatically by only sampling high-information areas around the particle56. This should allow tracking of the VLP all the way from the extracellular to the perinuclear space.
The skimming events observed here are to our knowledge the first of their kind and demonstrate how cell morphology can affect viral diffusion and how diffusion is inhibited at the cell surface. For membrane-bound VLPs, while the phenomenon of viruses utilizing actin-rich protrusions as tracks to facilitate transport along the plasma membrane has been well-described19,23,34,35,36,37,38,39, the distinct transport modes revealed here give insight into the different ways that VLPs interact with cellular protrusions. Finally, we demonstrated rapid diffusion of single VLPs in a tightly packed epithelial layer, which to our knowledge has never before been reported, paving the way toward high-speed SPT in more realistic biological systems. Future studies with 3D-TrIm will be able to probe various unanswered questions in viral dynamics, including the ‘molecular walker’ hypothesis57 and the seeming impenetrability of the periciliary layer for particles larger than 40 nm in diameter53. This work is also readily extendable to the study of pseudotyped SARS-CoV-2 VLPs, which have already been shown to mimic many of the properties of the wild-type virion58. Importantly, the application of this technique can be extended to any system where fast dynamics of nanoscale objects occur over large volumetric scales, including delivery of nanoscale drug candidates to the lungs59 and through leaky tumor vasculature60.
Methods
3D-TrIm instrument overview
The 3D-TrIm instrument design is shown in Extended Data Fig. 1 and consists of 3D-SMART tracking excitation optics and 3D-FASTR imaging excitation optics coupled through a commercial confocal microscope (Zeiss LSM 410, modified by LSM Tech), piezoelectric stage and microscope objective to join both setups together. The microscope is controlled by custom code LabVIEW code.
3D-SMART
High-speed particle tracking is achieved using a rapidly scanning laser focus over a narrow field of view and single-photon counting detectors to calculate photon-by-photon position updates in real time. This is accomplished using a pair of EODs to scan the laser across a 25-point Knight’s tour pattern44,61, covering a 1-µm2 area in the XY plane, holding at each spot for 20 µs (Fig. 1b). A TAG lens62 sinusoidally deflects the laser’s focus at ~70 kHz over a 2-µm range around the focal volume (Fig. 1b). Photon arrivals at different laser positions are fed into a Kalman filter63 on a field-programmable gate array. The real-time localization is then used as the input to an integral-feedback controller, which controls a piezoelectric nanopositioner to recenter the particle in the objective focal volume.
3D-SMART excitation optics
The optical pathway for 3D-SMART tracking excitation uses a continuous-wave solid-state 488-nm laser source (FCD488-30, JDSU) which is expanded 1.25× and spatially filtered with a 75-µm pinhole. The linear polarization of the beam is selected by a Glan–Thompson polarizer (GTH5-A, Thorlabs) and rotated by a half-wave plate (WPH05M-488, Thorlabs). A pair of EODs then deflect the polarized beam with a second half-wave plate in between to rotate the polarization orthogonal to the input. The deflected beam is further expanded 3.33× before focal modulation through an acoustic lens (TAG lens). This modulated focus is then relayed to the sample using three lenses which will preserve the input magnification. The beam is reflected into the underside of the LSM by a multiband dichroic mirror (DCM2) (Chroma ZT405/488/635rpc) and then reflected vertically toward the objective, collimated through a 200-mm lens (LA1708-A) situated in the underside of a slider containing a 700-nm shortpass dichroic (DCM1). The beam exits the slider and is focused by the objective lens.
3D-FASTR overview
In conventional laser scanning microscopes, a laser beam is raster-scanned point by point to form an image frame. Extension into the third dimension is achieved by translating the sample and performing serial frame acquisition at different depths. As the stage is used for active-feedback tracking of the particle, we instead used an ETL to create a continuous optical translation of the focus to provide an imaging depth range of up to 8 μm. This focal sweep completes several periods within the period of the frame raster scan, creating scan patterns across the Y–Z image plane which vary each frame-time. When the relative frequency between the ETL and the 2D frame scan are timed correctly (Supplementary Fig. 11f,g), a unique subset of voxel locations in the volume space are scanned each frame before the pattern repeats (Fig. 1c). This method, called 3D-FASTR, increases the volumetric imaging rate 4–8× beyond that provided by conventional volumetric microscopy methods because the resulting gaps between scanned voxels can be interpolated46. The 1-µs dwell time is common to most existing laser scanning microscopes and generates more than enough signal-to-noise to capture the morphology and dynamics of fluorescently labeled live cells.
The development of 3D-FASTR resulted in an imaging system capable of 3D imaging in motion, but since its original publication, the optical configuration of 3D-FASTR was re-designed to obtain a constant, diffraction-limited point-spread function throughout the focal range. These optical improvements also resulted in a triangular focal translation without the need for arbitrary drive signals. Improvements to 3D-FASTR optical performance are discussed in Supplementary Information Sections 1.2 and 2.1.
3D-FASTR excitation optics
Two-photon excitation was enabled by incorporating a tunable-wavelength (tuned to 800 nm), pulsed Yb 100-fs, 80-MHz laser source (Chameleon Discovery, Coherent) and expanded 2.5× using a Galilean telescope (ACN254-050-B, Thorlabs; AC254-125-B-ML, Thorlabs). Variable focusing of the expanded beam was achieved using an ETL (EL-10-30-C-NIR-LD-MV, Optotune) placed approximately 1,200 mm behind the rear aperture of the LSM System. A 450-mm lens (IL1) (11665, Edmund Optics) positioned 935 mm after the ETL enabled nearly linear focal translation with respect to current (Supplementary Fig. 11d), eliminating the need for any external modulation described in ref. 46. After passing through the confocal scan unit, the beam’s focal range is adjusted by a 1,000-mm lens (IL2) (LA1464-B, Thorlabs) placed on the imaging excitation side of the slider. The beam is then reflected vertically off a 700-nm shortpass dichroic mirror (DCM1) to the rear aperture of the objective lens (×100 Plan-Apo 1.4 NA, Zeiss).
Imaging variable focus detection
The beam is sampled before variable focusing using a 10:90 beamsplitter (BS025, Thorlabs). The sampled beam is focused using a 75-mm lens (AC254-075-B-ML, Thorlabs) onto a Si photodiode (DET10A, Thorlabs) terminated with a 56-kΩ resistor. This photodiode is used as a power reference to normalize the signal. Two additional 10:90 beamsplitters (BS1, BS2) sample the beam after focal modulation at extrema of the ETL focal range, producing peak photodiode signals at focal length minima (PD1) and maxima (PD2). Because the two measurement photodiode signals peak out-of-phase with respect to focal shift, their difference produces a monotonic response curve with respect to ETL focal power as reported previously in ref. 46. Details of the ETL focal length calibration can be found in Supplementary Information Section 1.2.
Shared emission pathway
A key factor to integrating the tracking and imaging modules in 3D-TrIm is minimization of crosstalk between the channels. In this implementation, tracking fluorescence collection occurs in the 500–600-nm window and imaging fluorescence is collected in the 600–700-nm window.
Emitted fluorescence from both tracking and imaging passes back through the dichroic mirror (DCM1) and 200-mm lens on the tracking excitation side of the slider. Both emitted beams pass through the multiband entrance dichroic (DCM2) and focus through an 85-mm lens positioned approximately 360 mm from the objective lens. Reflected imaging laser excitation is removed through a multiphoton blocking filter (FF01-750/SP-25, Semrock), and the tracking and imaging emissions are separated with a second dichroic mirror (DCM3) (Di03-R594-t1, Semrock). The imaging emission is focused onto a nondescanned detector (PMT) using a 50-mm lens (LA1131-A, Thorlabs) positioned 50 mm beyond the previous 85-mm lens.
After DCM3, the tracking emission is relayed by a 300-mm lens (AC254-300-A-ML, Thorlabs), placed 625 mm beyond the previous 85-mm lens, and focused onto an APD by an 80-mm lens (AC254-080-A-ML, Thorlabs). This detection setup results in a beam magnification that nearly fills the APD sensor area and prevents interference from the scanned imaging laser.
Tracking and imaging integration overview
The 3D-SMART active-feedback tracking and 3D-FASTR imaging are coupled into a single platform through the chromatically separated excitation and emission pathways described above but unified by the master spatial grid of the piezoelectric stage. The stage coordinates define the physical space and can be used as an absolute reference between tracking and imaging spaces. As the 3D-SMART system natively outputs the trajectory in stage-space coordinates, only the location of the 3D-FASTR image must be adjusted to generate fully registered tracking and imaging volume spaces. Multiple calibrations were used to obtain the imaging parameters needed to convert voxel locations from image-space into physical stage-space and are described below. Details for calibration of the relative tracking and imaging center positions can be found in Supplementary Information Section 1.1.
TrIm data acquisition workflow
Tracking and imaging acquisition is initiated by the 3D-SMART laser pattern scanning through the tracking volume. When the number of detected photons exceeds a predetermined threshold based on the laser power and observed background, a particle is assumed to be located within the tracking volume. This particle-detection event will initiate the stage feedback loop, open the imaging laser shutter and trigger data recording. Tracking and imaging data continue to be recorded until the tracking intensity drops below a threshold value based on the background level.
We note here that the primary limit to trajectory duration in diffusive scenarios is not photobleaching, but that trajectories most frequently ended when reaching the boundary of the piezoelectric stage. For confined trajectories, acquisition continues until bleaching causes the virus intensity to fall to a level similar to the background.
Automated acquisition overview
Due to the serial nature of SPT acquisition, obtaining a quantity of trajectories sufficient for population-level statistical analysis can be tedious and time-consuming. We developed an automated acquisition protocol to act as an autopilot so that 3D-TrIm experiments could be conducted without user input over experiment durations up to 12 h. The cells were maintained at 37 °C and buffered (Live Cell Imaging Solution, Invitrogen A14291DJ) with 2% FBS (Millipore Sigma, no. F2442) to preserve viability through the duration of the experiment.
The autopilot algorithm searches for particles and considers any particle-detection event as a trajectory regardless of duration. After a set duration, either time- or quantity-based, an area-migration event is triggered, which moves the motorized stage to a new location in an S pattern to minimize overall distance traveled. The algorithm then continues to search for particles in the new area and repeats this process until it is manually terminated, or an imposed time limit has been reached. A typical autopilot session results in acquisition of ~500 trajectories.
TrIm data processing and visualization workflow
The goal of data processing in 3D-TrIm is to register tracking and imaging datasets in the same stage-space, which requires conversion of image data from pixel-space to stage-space. The pixel-based nature of 3D-FASTR acquisition results in image data without fixed temporal boundaries: any number of 3D volumes can be flexibly created from the total acquired image data, limited only by the desired balance between image quality and speed.
In the static-stage 3D-FASTR imaging case, the precise timing of the relative frame and ETL frequencies enables the out-of-sequence sampling to scan a volume completely without multiply sampling a voxel location more than once. The theoretical time to scan a complete volume, Tvol, is equivalent to the product of the frame-time and number of z-slices in the volume. In 3D-TrIm, however, the motion of the stage across Z expands the volume beyond the initial size given by the ETL depth range alone. We observed axial diffusion ranges of up to 25 µm in a single Tvol period. To account for increased volume size, we use the static-case Tvol value as our base volume acquisition time.
Once the temporal volume boundaries are determined, the voxel positions are transformed from image frame position (Pimage) into the stage coordinate space position (Vrel) in each axis using the calibrated value for the trackcenter (Pcenter) and the pixel size (Ψ, µm per pixel) according to the equation below, which shows this transformation for the x-axis positions.
This value is then offset by the position of the stage, S, to yield the absolute position in physical space \((V_{X,{\mathrm{abs.}}})\).
The sign is determined by the relative orientation between the piezo stage and laser scan as obtained through the slope of the pixel size calibration described above and in Supplementary Information Section 1.3.
This process is simplified for the z axis because the ETL calibration is zero-referenced to the axial trackcenter in micrometers, such that only the stage offset in equation 2 is necessary for voxel placement. After conversion, the stage-space voxel positions must be re-discretized to form the global voxel grid. This is accomplished by using the stage-space extrema as the centers of the resulting minimum and maximum voxels. The locations of these voxel boundaries are determined per trajectory based on the size of the global volume space.
After voxel assignment, each local volume is assessed for sampling to prevent overinterpolation of undersampled image planes and cropped such that the exterior planes of the volume are at least 20% sampled. The MATLAB (MathWorks) function inpaintn64 is used to perform the inpainting operation. After interpolation, the local volume intensity is rescaled by setting the saturation point at the intensity equal to the cumulative distribution threshold of 0.9998 and contrast-enhanced by scaling with a gamma value of 0.7. Unless a time series is desired, these completed local volumes can be composited into a single maximum intensity projection of the global volume over time. When displayed as a time series of local volumes, the image may appear incomplete or irregularly stripy in some regions. This occurs when the virion diffuses toward or away from the cells in Z such that cells are visible in the imaging volume for only a portion of the local volume time. Compositing into a global volume is recommended for forming distance maps to avoid such issues.
Preparation of fluorescently labeled VLPs
For these single virus tracking experiments, we incorporated fluorescent protein into VSV-G VLPs by fusing either eGFP, or miRFP670, to HIV-1 Vpr, which is packaged within the pseudovirus nucleocapsid. 3D-TrIm trajectories were acquired on cells labeled with SYTO61 (targeted to nucleic acids) or SiR650-actin (targeted to F-actin). The cell labels were chosen to maximize chromatic separation between the tracking and imaging, and the largest contributor to tracking crosstalk was cell autofluorescence by single-photon excitation, which was low enough not to perturb the active-feedback single-virus tracking. We cultured monolayers of HeLa, BJ fibroblasts or LDLR-deficient GM701 fibroblasts on glass coverslips. Alternatively, to achieve multi-layered cells, we cultured HT29-MTX cells on inverted matrix support filters65. This selection of cell types offered diversity in morphology, extracellular environment and cell-surface receptor concentration to observe their influences on the early stages of viral contacts.
Plasmid construction
The expression vector eGFP-Vpr, used to generate internally labeled VSV-G VLPs, was constructed as follows. The vector pVpr+ backbone was derived from mCherry-2xCL-YFP-Vpr (Addgene, no. 105215) and amplified by PCR with the following primers: 5′-TTTTCCTGCAGGTGAACAAGCCCCAGAAGACC-3′ (SbfI site underlined) and 5′-TTTTACCGGTCGTCGACTGCAGAATTCG-3′. DNA encoding for the fluorescent protein eGFP was amplified by PCR with the following primers: 5′-TTGCTAGCGCTACCGGTC-3′ (NheI site underlined) and 5′-TCCGCCTGCAGGCTTGTACAGCTCGTCCATG-3′ (SbfI site underlined), using eGFP-Rab7 (Addgene, no. 12605) as the template. Both PCR products were digested with NheI + SbfI and ligated together to create eGFP-Vpr; successful insertion was verified by sequencing.
The expression vector pmiRFP70.Vpr, used to generate internally labeled VSV-G VLPs, was derived from the same backbone as above and amplified by PCR with the following primers: 5′-TTTTACCGGTCGTCGACTGCAGAATTCG-3′ (AgeI site underlined) and 5′-AAAACCTGCAGGTGAACAAGCCCCAGAAGAC-3′ (SbfI site underlined). DNA encoding for the fluorescent protein miRFP670 was amplified by PCR with the following primers: 5′-GATCCACCGGTCGCC-3′ (AgeI site underlined) and 5′-TTTTCCTGCAGGGCTCTCAAGCGCGG-3′ (SbfI site underlined), using pmiRFP670-N1 (Addgene, no. 79987) as the template. Both PCR products were digested with AgeI + SbfI and ligated together to create pmiRFP670-Vpr; successful insertion was verified by sequencing.
Production of VSV-G VLP containing eGFP-Vpr
Viral vectors were produced by the Duke Viral Vector Core Facility (Department of Neurobiology, Duke University School of Medicine), as described previously66. Briefly, HEK-293T cells grown in 10-cm plates were transfected using a calcium phosphate-based protocol with either 10 µg of psPAX2, 2.5 µg of pREV, 5 µg pMD2.G (VSV-G) and 5 µg of the eGFP-Vpr plasmid (described above), or 10 µg of psPAX2, 2.5 µg of pREV, 5 µg pMD2.G (VSV-G), 15 µg of the transgene/reporter gene pLenti-GFP and 5 µg of the pmiRFP670.Vpr plasmid (described above), to produce VLPs suitable for SPT or transduction assays, respectively. In both cases, the medium was replaced at 24 h after transfection, and at 72 h posttransfection, supernatant containing pseudotyped particles was centrifuged at 450g for 10 min and filtered through a 0.45-μm pore-size filter.
Viral titers were determined using a p24 ELISA and reported in transducing units per ml (TU ml−1), typically 2–4 × 107 TU ml−1. The virus solution was stored in aliquots at −80 °C. Before SPT and immunofluorescence experiments, the virus solution was buffer exchanged by dialysis (Spectra-Por Float-A-Lyzer G2, MWCO 100 kDa, Millipore Sigma) against PBS (Genesee Scientific, Cat. no. 25-507) at 4 °C.
Cell culture
HeLa cells (Duke Cell Culture Facility, ATCC. no. CRL-1958), fibroblasts (Duke Cell Culture Facility, BJ, ATCC, no. CRL-2522), hypercholesterolemia fibroblasts (Coriell Institute, GM00701) and mucus-producing differentiated goblet cells (HT29-MTX-E12, Millipore Sigma, no. 12040401) were all grown using complete DMEM which comprised DMEM (Corning, no. 10-013-CV) supplemented with 10% FBS (Millipore Sigma, no. F2442) and 1 × penicillin-streptomycin (Corning, no. 30-002-CI). Cells were maintained at 37 °C with 5% CO2 and passaged when they reached ~70% confluency.
Cell staining and microscope sample preparation
At 24 h before microscopy, HeLa and fibroblast cells were plated in complete DMEM at 8 × 105 cells per well in a 6-well plate with an autoclaved glass coverslip (VWR, no. CLS-1760-025) to ensure 80% confluency on the day of the experiment. HT29-MTX cells were seeded on the underside of a Transwell permeable filter (Millipore Sigma, no. CLS3460) in complete DMEM, which after attachment (4 h at 37 °C and 5% CO2) was reinverted and cultured for 72 h before microscopy65. Cells were stained with either nucleic acid stain (SYTO61, ThermoFisher, no. S11343) or F-actin stain (SiR-actin, Siprochrome, no. SC001). Cells were incubated with 2 µM SYTO61 in complete DMEM for 30 min at 37 °C and 5% CO2 followed by 3× wash steps (1 × PBS). Alternatively, cells were incubated with 250–100-nm SiR-actin in complete DMEM for 6–14 h at 37 °C and 5% CO2 followed by 3× wash steps (1 × PBS). In each case, after staining the coverslip was transferred to HEPES pH 7.4 buffered solution (live cell imaging solution, ThermoFisher, no. A14291DJ) in a custom-built sample holder and positioned on a heated stage (37 °C). All experiments were performed using live cells.
Live cell volumetric imaging and real-time viral tracking were performed on cells prepared as described above. eGFP-Vpr VLP was added to cells on a heated stage to initiate the reaction to a final concentration of 1.9 × 106 TU ml−1, which approximates to multiplicity of infection ~1. The average excitation power at the focus was 180 nW and 7.5 mW for the 488-nm and 800-nm beams, respectively.
Statistics and reproducibility
Over 3,000 single virus trajectories were collected over the course of the study, taken from more than 21 individual experimental runs. Numbers of independent viral trajectories for all statistical analyses are reported in the related figure captions. Statistical significance of differences between distributions were calculated using Kruskal–Wallis or t-tests. No statistical method was used to predetermine sample sizes. No data were excluded from the analyses. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Raw data files for 3D-TrIm trajectories in all main text figures can be found at the Duke University Research Data Repository at https://doi.org/10.7924/r4bp07h15.
Code availability
Code to analyze single virus trajectories (MATLAB) and render them (Amira) is available at https://github.com/welsherlab/3dtrim.
References
Baos, S. C., Phillips, D. B., Wildling, L., McMaster, T. J. & Berry, M. Distribution of sialic acids on mucins and gels: a defense mechanism. Biophys. J. 102, 176–184 (2012).
Kesimer, M. et al. Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol. 6, 379–392 (2013).
Linden, S. K., Sutton, P., Karlsson, N. G., Korolik, V. & McGuckin, M. A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 1, 183–197 (2008).
Ridley, C. & Thornton, D. J. Mucins: the frontline defence of the lung. Biochem. Soc. Trans. 46, 1099–1106 (2018).
Thornton, D. J., Rousseau, K. & McGuckin, M. A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 70, 459–486 (2008).
Tomlin, H. & Piccinini, A. M. A complex interplay between the extracellular matrix and the innate immune response to microbial pathogens. Immunology 155, 186–201 (2018).
Pais-Correia, A.-M. et al. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 16, 83–89 (2010).
Brandenburg, B. & Zhuang, X. Virus trafficking—learning from single-virus tracking. Nat. Rev. Microbiol. 5, 197–208 (2007).
Liu, S. L. et al. Single-virus tracking: from imaging methodologies to virological applications. Chem. Rev. 120, 1936–1979 (2020).
Parveen, N., Borrenberghs, D., Rocha, S. & Hendrix, J. Single viruses on the fluorescence microscope: imaging molecular mobility, interactions and structure sheds new light on viral replication. Viruses 10, 250 (2018).
Bhagwat, A. R. et al. Quantitative live cell imaging reveals influenza virus manipulation of Rab11A transport through reduced dynein association. Nat. Commun. 11, 23 (2020).
Lakdawala, S. S. et al. Influenza A virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 10, e1003971 (2014).
Endress, T. et al. HIV-1-cellular interactions analyzed by single virus tracing. Eur. Biophys. J. 37, 1291–1301 (2008).
Seisenberger, G. et al. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 294, 1929–1932 (2001).
Rust, M. J., Lakadamyali, M., Zhang, F. & Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 11, 567–573 (2004).
Pelkmans, L., Kartenbeck, J. & Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 3, 473–483 (2001).
Cureton, D. K., Harbison, C. E., Cocucci, E., Parrish, C. R. & Kirchhausen, T. Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. J. Virol. 86, 5330–5340 (2012).
Sun, E. Z. et al. Real-time dissection of distinct dynamin-dependent endocytic routes of influenza A virus by quantum dot-based single-virus tracking. ACS Nano 11, 4395–4406 (2017).
Xu, H. et al. Real-time imaging of rabies virus entry into living vero cells. Sci. Rep. 5, 11753 (2015).
Lakadamyali, M., Rust, M. J., Babcock, H. P. & Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl Acad. Sci. USA 100, 9280–9285 (2003).
van der Schaar, H. M. et al. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 4, e1000244 (2008).
van der Schaar, H. M. et al. Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol. 81, 12019–12028 (2007).
Coller, K. E. et al. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 5, e1000702 (2009).
Qin, C. et al. Real-time dissection of dynamic uncoating of individual influenza viruses. Proc. Natl Acad. Sci. USA 116, 2577–2582 (2019).
Li, Q. et al. Single-particle tracking of human immunodeficiency virus type 1 productive entry into human primary macrophages. ACS Nano 11, 3890–3903 (2017).
Ewers, H. et al. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl Acad. Sci. USA 102, 15110–15115 (2005).
Suomalainen, M. et al. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144, 657–672 (1999).
McDonald, D. et al. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452 (2002).
Bailey, C. J., Crystal, R. G. & Leopold, P. L. Association of adenovirus with the microtubule organizing center. J. Virol. 77, 13275–13287 (2003).
Arhel, N. et al. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 3, 817–824 (2006).
Liu, S. L. et al. Effectively and efficiently dissecting the infection of influenza virus by quantum-dot-based single-particle tracking. ACS Nano 6, 141–150 (2012).
Zhang, L. J. et al. A ‘driver switchover’ mechanism of influenza virus transport from microfilaments to microtubules. ACS Nano 12, 474–484 (2018).
Wu, Q. M. et al. Uncovering the Rab5-independent autophagic trafficking of influenza A virus by quantum-dot-based single-virus tracking. Small 14, e1702841 (2018).
Sherer, N. M. et al. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 9, 310–315 (2007).
Dixit, R., Tiwari, V. & Shukla, D. Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci. Lett. 440, 113–118 (2008).
Schelhaas, M. et al. Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 4, e1000148 (2008).
Sowinski, S. et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10, 211–219 (2008).
Mercer, J. & Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320, 531–535 (2008).
Jose, J., Tang, J., Taylor, A. B., Baker, T. S. & Kuhn, R. J. Fluorescent protein-tagged Sindbis virus E2 glycoprotein allows single particle analysis of virus budding from live cells. Viruses 7, 6182–6199 (2015).
Gao, L., Shao, L., Chen, B.-C. & Betzig, E. 3D live fluorescence imaging of cellular dynamics using Bessel beam plane illumination microscopy. Nat. Protoc. 9, 1083–1101 (2014).
Planchon, T. A. et al. Rapid three-dimensional isotropic imaging of living cells using Bessel beam plane illumination. Nat. Methods 8, 417 (2011).
Hou, S., Johnson, C. & Welsher, K. Real-time 3D single particle tracking: towards active feedback single molecule spectroscopy in live cells. Molecules 24, 2826 (2019).
Hou, S., Exell, J. & Welsher, K. Real-time 3D single molecule tracking. Nat. Commun. 11, 3607 (2020).
Hou, S., Lang, X. & Welsher, K. Robust real-time 3D single-particle tracking using a dynamically moving laser spot. Opt. Lett. 42, 2390–2393 (2017).
Hou, S. & Welsher, K. An adaptive real-time 3D single particle tracking method for monitoring viral first contacts. Small 15, e1903039 (2019).
Johnson, C., Exell, J., Kuo, J. & Welsher, K. Continuous focal translation enhances rate of point-scan volumetric microscopy. Opt. Express 27, 36241–36258 (2019).
Finkelshtein, D., Werman, A., Novick, D., Barak, S. & Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl Acad. Sci. USA 110, 7306–7311 (2013).
Naldini, L. et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 (1996).
Desai, T. M. et al. Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 12, 88 (2015).
Montiel, D., Cang, H. & Yang, H. Quantitative characterization of changes in dynamical behavior for single-particle tracking studies. J. Phys. Chem. B 110, 19763–19770 (2006).
Welsher, K. & Yang, H. Multi-resolution 3D visualization of the early stages of cellular uptake of peptide-coated nanoparticles. Nat. Nanotechnol. 9, 198–203 (2014).
Jensen, C. & Teng, Y. Is it time to start transitioning from 2D to 3D cell culture? Front. Mol. Biosci. 7, 33 (2020).
Button, B. et al. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337, 937–941 (2012).
Perillo, E. P. et al. Deep and high-resolution three-dimensional tracking of single particles using nonlinear and multiplexed illumination. Nat. Commun. 6, 7874 (2015).
Nasir, W., Bally, M., Zhdanov, V. P., Larson, G. & Höök, F. Interaction of virus-like particles with vesicles containing glycolipids: kinetics of detachment. J. Phys. Chem. B 119, 11466–11472 (2015).
Zhang, C. & Welsher, K. Information-efficient, off-center sampling results in improved precision in 3D single-particle tracking microscopy. Entropy (Basel) 23, 498 (2021).
Hamming, P. H. E., Overeem, N. J. & Huskens, J. Influenza as a molecular walker. Chem. Sci. 11, 27–36 (2020).
Peacock, T. P. et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 6, 899–909 (2021).
Bobba, C. M. et al. Nanoparticle delivery of microRNA-146a regulates mechanotransduction in lung macrophages and mitigates injury during mechanical ventilation. Nat. Commun. 12, 289 (2021).
Sun, D., Zhou, S. & Gao, W. What went wrong with anticancer nanomedicine design and how to make it right. ACS Nano 14, 12281–12290 (2020).
Wang, Q. & Moerner, W. E. Optimal strategy for trapping single fluorescent molecules in solution using the ABEL trap. Appl. Phys. B 99, 23–30 (2010).
Mermillod-Blondin, A., McLeod, E. & Arnold, C. B. High-speed varifocal imaging with a tunable acoustic gradient index of refraction lens. Opt. Lett. 33, 2146–2148 (2008).
Fields, A. P. & Cohen, A. E. Optimal tracking of a Brownian particle. Opt. Express 20, 22585–22601 (2012).
Garcia, D. Robust smoothing of gridded data in one and higher dimensions with missing values. Comput. Stat. Data Anal. 54, 1167–1178 (2010).
Wakabayashi, Y., Chua, J., Larkin, J. M., Lippincott-Schwartz, J. & Arias, I. M. Four-dimensional imaging of filter-grown polarized epithelial cells. Histochem. Cell Biol. 127, 463–472 (2007).
Ortinski, P. I., O’Donovan, B., Dong, X. & Kantor, B. Integrase-deficient lentiviral vector as an all-in-one platform for highly efficient CRISPR/Cas9-mediated gene editing. Mol. Ther. Methods Clin. Dev. 5, 153–164 (2017).
Acknowledgements
We acknowledge financial support from the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM124868 (K.D.W.). We also acknowledge the Duke Viral Vector Core for assistance with virus-like particle generation and the Duke Cell Culture Facility for access to cell lines used in this study. We further thank LSM Tech for help customizing the laser scanning microscope for integration into the 3D-TrIm setup. We are also grateful to Duke Office of Information Technology (OIT) and Duke Research Computing for facilitating access to Amira 3D 2021.1 for data rendering.
Author information
Authors and Affiliations
Contributions
C.J., J.E. and K.D.W. conceptualized the study. C.J., J.E. and K.D.W. were responsible for the methodology. C.J., J.E. and Y.L. performed investigations. C.J., J.E., Y.L., J.A. and K.D.W. curated the data. C.J., J.E., Y.L., J.A. and K.D.W. performed visualizations. K.D.W. was responsible for funding acquisition. C.J., J.E. and K.D.W. wrote the original draft and reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Hari Shroff and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Madhura Mukhopadhyay, in collaboration with the Nature Methods team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Instrument Diagram.
3D-TrIm Instrument Diagram. The 3D-TrIm microscope consists of two excitation sources: one for single-particle tracking and one for imaging. The tracking laser position is modulated using two electro-optic deflectors (EOD) and tunable acoustic gradient (TAG) lens. The imaging laser focus is modulated by an electrically tunable lens (ETL) and the beam is sampled by beam-splitters (BS) and evaluated using a system of photodiodes (PD). This focus is relayed using two lenses (IL). The tracking laser enters the underside of the LSM410 through a dichroic mirror (DCM2) where it ultimately couples with the imaging laser through DCM1 toward the shared objective lens and piezoelectric stage. After exciting the sample, the fluorescence emission pathway is shared between tracking and imaging until separated by DCM3.
Extended Data Fig. 2 Comparison of image-based tracking and imaging (spinning disk confocal) with active-feedback tracking and complementary 3D imaging (3D-TrIm).
Fluorescently-labeled virus-like particles (eGFP-Vpr VSVG) were introduced to cultures of live HeLa cells stained with SiR650-actin (100 nm). a-e, Data collected on Andor Dragon fly spinning disk confocal microscope. The same area was sampled continuously. Each volume has a depth of 8 μm split into 16 z planes. Virus particles and cells were imaged simultaneously with a camera exposure time of 40 msec. f-j, Data collected on 3D-TrIm microscope. A single virus particle was tracked continuously (1 msec sampling shown) and the surrounding area was imaged 5 μm below the particle and 3 μm above, to give an approximate 8 μm volume. The resulting trajectories over the entire acquisition period are shown and color-coded by time. Gray box in XZ projections shows the size of the spinning disk imaging volume for comparison. Experiments were performed in live-cell image solution with 2% FBS and maintained at 37 °C.
Extended Data Fig. 3 High resolution tracking and 3D imaging data, related to Fig. 2.
a, Local Volume MIPs. b, Tracking intensity trace. Both a and b are associated with VSV-G/HeLa tracking and imaging render, Fig. 2c–g. c, Local Volume MIPs, local volumes from 138-162 sec have been omitted as trajectory diffuses a greater distance above the cells than the axial range of the ETL. Note in volume 2 from 17-34s there is a noted absence of intensity due to transit in Z of the particle away from the cells. This can appear as striping due to unsampled voxels and is a feature of local volumes, especially when displayed as MIPs. d, Tracking intensity trace. Both c and d are associated VSV-G/GM701 tracking and imaging render, Fig. 2h-j.
Extended Data Fig. 4 Virus-to-cell distance map calculation.
a and b, The full trajectory is shown in white. Using the top-hat-generated isosurface (gray, see Supplementary Information 3.4) from 3D-FASTR imaging data the distance from each trajectory point to each surface vertex is calculated as describe in Supplementary Information Section 3.5. The minimum distance is then assigned to each data point (purple lines, only every 50th data point is shown for clarity and every 10th data point in the insert). The vectors from the vertex to the data point corresponding to the minimum distance are also shown as spheres on the cellular contour. a is related to trajectory shown in Fig. 2c-g, b is related to trajectory shown in Fig. 2h-j.
Extended Data Fig. 5 Additional example 1 of VSV-G VLP freely diffusing in the extracellular space.
a, Top-down view of virus trajectory pincering live HeLa cell. b, Virus-to-cell distance representation of trajectory in a. a and b share the same axes and scale bar. c,d, Different view of the trajectory shown in a, b. Circular insets shows close-up views of virus in close contact with the cell surface. c and d share the same axes and scale bar. e, Cross-section with slice along x and y axis, overlaid with trajectory color-coded by diffusion coefficient. a, c, and e are color-coded by image intensity, while b and d display virus-to-cell distance map on surface of cell. f, Top, correlation between diffusivity and distance from the cell surface of VSV-G VLP, close-approaches (below 0.5 µm) are color patched purple. Dashed purple line, 0.5 µm from cell surface. Below left, enlarged view of data between 14.5-25.5 sec, associated with trajectory segment displayed in circular inset of d. Below right, enlarged view of data between 43.25-56 sec, associated with trajectory segment displayed in circular inset of d. g, Visual representation of virus-to-cell distance calculation. Isosurface volume render of cell image (gray) overlayed with trajectory sampled at 3 msec (white). Distance vectors are calculated from each trajectory timepoint to the isosurface (generated by top-hat transform) and displayed as color-coded spheres on the cell surface.
Extended Data Fig. 6 Additional example of VSV-G VLP Binding Event.
a, 3D-TrIm render of BJ Fibroblast cells stained with SiR-650 (f-Actin). b, Distance-diffusivity trace of viral trajectory. This trajectory begins with rapid extracellular diffusion before binding to the surface. A small ~1 second detachment occurs before re-binding for several seconds and detaching again where skimming is observed with inhibited diffusivity compared to the initial free diffusion, followed by free diffusion that has greater diffusivity than observed while skimming, but lower than the initial free diffusion.
Extended Data Fig. 7 Additional example 1 of VLP-protrusion interaction.
a, Top-down view of VSV-G VLP landing on protrusion from the surface of a live cell. 3D volume rendering of live HeLa cells (stained with SYTO61) co-registered with high resolution virus trajectory from 4D tracking and imaging data. b, c Alternate views of the trajectory shown in a. d, Magnified views showing cylindrical nature of trajectory and proximity to cell surface. e, Top-down view, with trajectory colored by diffusion coefficient, free portion with D > 1.5 × 10−2 μm2/s colored gray. f, Color-coded trajectory, showing free diffusion (D > 1.5 × 10−2 μm2/s, gray) and bound diffusion (green-yellow). g, Cylindrical fitting of the bound portion of the trajectory in a-f (28-120 sec), fitted cylinder (magenta) has a radius of 122 ± 8.4 nm after taking into consideration the tracked particle diameter. h, View of trajectory along the cylinder axis. i, Intensity trace of tracked VLP.
Extended Data Fig. 8 High resolution tracking and simultaneous 3D imaging localizes VSV-G VLP diffusion along fibroblast protrusion.
a, Left, top-down view of VSV-G VLP ‘surfing’ on actin-stained protrusion of BJ fibroblast. Trajectory colored by time. The lower, brighter, z-planes have been omitted to help visualize the protrusion the VLP is localized to. Right, same view but trajectory color-coded by diffusivity. b, Snapshots of 3D reconstruction. The sustained linear motion correlates with the lateral surface of the protrusion, large changes in diffusivity where detected. c, (left) XY MIP, (middle) magnified, and (right) YZ MIP of viral diffusion on actin-rich protrusion.
Extended Data Fig. 9 Additional example of VSV-G VLP circumnavigating HT29-MTX cells.
a, 3D reconstruction from a 4D data set covering 3 local volumes, at 16 FPV of suspended HT29-MTX cells grown on inverted matrix stained with SiR650-actin with high resolution trajectory. Below, axial view. b, Magnified view. c, Cell boundary reconstruction highlighting sample density. d, XY MIP. e, 3D reconstruction of cellular imaging data in a with trajectory split into unique segments of diffusion behavior via change-point analysis. f, VLP intensity trace.
Extended Data Fig. 10 Multi-trajectory acquisition.
a, Left: 10 VSV-G VLP trajectories collected in the same area of live HeLa cells stained with SYTO61 (see also: Supplementary Fig. 29 and Supplementary Video 8). Right: top-down (xy) MIP with multiple co-registered VLP trajectories. Colorbar represents trajectory number, with each trajectory uniquely colored. b, The dwell time of 27 VSV-G VLP trajectories (starting from free diffusion) collected in the same area as a were binned into voxels and represented as spheres, color-mapped by diffusivity. This correlated analysis reveals commonly visited regions and highlights the slower diffusion experienced by the VLP population nearer the cell surface. Inset: two different VLPs land in close proximity on the same cell. c, Exploded views showing multiple VLPs contact the same cell.
Supplementary information
Supplementary Information
Supplementary methods and Figs. 1–30, and description of Supplementary Videos.
Supplementary Video 1
3D-TrIm method animation, related to Fig. 1. Demonstration of 3D-TrIm operating principle. Animation sequence begins with overview of experimental setup in which a heated sample containing virus-like particles (VLP) and live cells are mounted on a piezoelectric stage with an objective lens shared by both tracking and imaging microscope sources. This overview is followed by an animation of 3D-SMART real-time tracking, demonstrating how a pair of Electro-Optic Deflectors (EOD) create a lateral Knight’s Tour grid pattern, followed by the use of a Tunable Acoustic Gradient (TAG lens) to scan a focal range above and below the center of the focal volume. A final animation demonstrates the principle of 3D-FASTR point-scan imaging.
Supplementary Video 2
VSV-G exploring the extracellular matrix, related to Fig. 2a,b. 3D reconstruction of real-time VSV-G VLP trajectory in extracellular matrix of live GM701 cells (stained with F-actin label SiR650-actin), from a 4D dataset covering 10 local volumes, at 16 FPV. Trajectory (~162 s) is segmented into 25 segments per second (25 frames per second when playback rate is 1×) and color mapped by time. The progress bar shows how the trajectory is further categorized: (1) Free diffusion period (playback rate: 2×): 0–14 s, 18–38 s, 44–62 s, 70–108 s. (2) Skimming period (playback rate: 1×): 14–18 s, 38–44 s, 62–70 s, 108–122 s. (3) Detachment (playback rate: 2×): 122–162 s. Sphere represents the VLP position in the current frame (refreshing rate is consistent with the trajectory, that is, 25 FPS at 1× playback rate). Image volumes formed from maximum intensity projection over time from local volumes acquired over 16 frame-times. In a, cells are color-coded by imaging intensity, while in b, cells are color-coded depending on distance of the virus from the cell surface. Panels a and b share the same trajectory color scale, camera angle and camera path; however, a is magnified compared with b.
Supplementary Video 3
VSV-G VLP skimming and binding, related to Fig. 3a–c. 3D reconstruction of live HeLa cells (stained with nucleic acid label SYTO61) corendered with virus trajectory. Trajectory (~123 s) is segmented into 25 segments per second (25 frames per second when playback rate is 1×) and color mapped by time. The progress bar shows how the trajectory is further categorized: (1) Free diffusion period (playback rate: 2×): 14–30 s, 41–70 s. (2) Skimming period (playback rate: 1×): 0–14 s, 30–41 s. (3) Immobilized period (playback rate: 4×): 70–123 s. The end points of trajectory segments are labeled with spheres (refreshing rate is consistent with the trajectory, that is, 25 FPS at 1× playback rate), representing the position of the viral particle. The diffusional status of viral particle is further illustrated by the color change of the sphere (gray, free motion; cyan, skimming behavior; purple, immobilization). Image volumes formed from maximum intensity projection over time from local volumes acquired over 16 frame-times. Cells are color-coded by image intensity.
Supplementary Video 4
VSV-G VLP transient binding, related to Fig. 3e–g. 3D reconstruction of live GM701 cells (stained with F-actin label SiR650-actin) corendered with virus trajectory. Trajectory (~54 s) is segmented into 25 segments per second and color mapped by diffusivity. The playback rate is constant 1×. Image volumes formed from maximum intensity projection over time from local volumes acquired over 16 frame-times. Cells are color-coded by image intensity. We observed the VLP repeatedly landing and detaching.
Supplementary Video 5
VSV-G VLP interaction with cell surface protrusion, related to Fig. 4. 3D reconstruction of live HeLa cells (stained with nucleic acid label SYTO61) corendered with virus trajectory. Trajectory (~133 s) is segmented into 25 segments per second (25 frames per second when playback rate is 1×) and color mapped by time. Image volumes formed from maximum intensity projection over time from local volumes acquired over 16 frame-times. Cells are color-coded by image intensity. Shown in the progress bar, the trajectory is further categorized into two segments: (1) Free diffusion (playback rate: 1×): 0–16 s. (2) Membrane-bound diffusion on protrusion: 16–133 s.
Supplementary Video 6
VSV-G VLP diffusing through multi-layered epithelial cells, related to Fig. 5. 3D reconstruction of suspended HT29-MTX cells grown on inverted matrix support (stained with F-actin label SiR650-actin) corendered with virus trajectory. Trajectory (~166 s) is segmented into 25 segments per second (25 frames per second) and color mapped by time. The playback rate is constant 4×. This video shows how the global volume intensity is accumulated from local volumes acquired over four frame-times. Cells are color-coded by image intensity. Axes grid represents approximate position of matrix support on which the cells are suspended.
Supplementary Video 7
Internalized viral trafficking, related to Fig. 6. 3D reconstruction of live HeLa cells (stained with F-actin label SiR650) corendered with virus trajectory. Trajectory (~325 s) color mapped by time. Global image volume formed from maximum intensity projection over time from local volumes acquired over 16 frame-times. Cells are color-coded by image intensity. Initially, the cells are opaque and the trajectory is not visible; a cut-through reveals the complete internalized trajectory. Viewing this embedded trajectory from above reveals high-intensity, actin-rich regions which the trajectory appears to navigate. Finally, the camera is rotated to a lateral view to show the position of the trajectory within the 3D cell.
Supplementary Video 8
Multi-trajectory overlay, related to Extended Data Fig. 10. 3D reconstruction of a single assembled area containing live HeLa cells (stained with nucleic acid label SYTO61) corendered with ten virus trajectories. The total imaging data were acquired during the ten trajectories and coregistered to form a single common area. Image volumes formed from maximum intensity projection over time from the assembled volumes acquired over 16 frame-times. Cells are color-coded by image intensity. Ten trajectories combined (~490 s in total) are segmented into 25 segments per second (25 frames per second when playback rate is 1×). The trajectories are color mapped with respect to trajectory number (the color legend in lower left indicates the initial color of each trajectory). Each trajectory is further color mapped by time, as indicated in the progress bar. Trajectory no. 5 is related to Supplementary Video 3.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Johnson, C., Exell, J., Lin, Y. et al. Capturing the start point of the virus–cell interaction with high-speed 3D single-virus tracking. Nat Methods 19, 1642–1652 (2022). https://doi.org/10.1038/s41592-022-01672-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-022-01672-3
This article is cited by
-
A paintbrush for delivery of nanoparticles and molecules to live cells with precise spatiotemporal control
Nature Methods (2024)
-
Next-generation single virus tracking
Nature Methods (2022)
