
Abstract
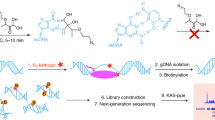
Transcription is a highly dynamic process that generates single-stranded DNA (ssDNA) in the genome as ‘transcription bubbles’. Here we describe a kethoxal-assisted single-stranded DNA sequencing (KAS-seq) approach, based on the fast and specific reaction between N3-kethoxal and guanines in ssDNA. KAS-seq allows rapid (within 5 min), sensitive and genome-wide capture and mapping of ssDNA produced by transcriptionally active RNA polymerases or other processes in situ using as few as 1,000 cells. KAS-seq enables definition of a group of enhancers that are single-stranded and enrich unique sequence motifs. These enhancers are associated with specific transcription-factor binding and exhibit more enhancer–promoter interactions than typical enhancers do. Under conditions that inhibit protein condensation, KAS-seq uncovers a rapid release of RNA polymerase II (Pol II) from a group of promoters. KAS-seq thus facilitates fast and accurate analysis of transcription dynamics and enhancer activities simultaneously in both low-input and high-throughput manner.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
All sequencing data are available at NCBI Gene Expression Omnibus with the accession number: GSE139420. Other data that support the findings of this study are available from the corresponding author upon request.
Change history
31 May 2020
A Correction to this paper has been published: https://doi.org/10.1038/s41592-020-0881-1
References
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Kim, T.-K. et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187 (2010).
Core, L. J., Waterfall, J. J. & Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848 (2008).
Kwak, H., Fuda, N. J., Core, L. J. & Lis, J. T. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953 (2013).
Fuchs, G. et al. 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol. 15, R69 (2014).
Schwalb, B. et al. TT-seq maps the human transient transcriptome. Science 352, 1225–1228 (2016).
Churchman, L. S. & Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373 (2011).
Christopher, M. W., Ramachandran, S. & Henikoff, S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 53, 819–830 (2014).
Nojima, T. et al. Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161, 526–540 (2015).
Mayer, A. et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161, 541–554 (2015).
Hirabayashi, S. et al. NET-CAGE characterizes the dynamics and topology of human transcribed cis-regulatory elements. Nat. Genet. 51, 1369–1379 (2019).
Mirkovitch, J. & Darnell, J. E. Mapping of RNA polymerase on mammalian genes in cells and nuclei. Mol. Biol. Cell 3, 1085–1094 (1992).
Muse, G. W. et al. RNA polymerase is poised for activation across the genome. Nat. Genet. 39, 1507–1511 (2007).
Kouzine, F. et al. Global regulation of promoter melting in naive lymphocytes. Cell 153, 988–999 (2013).
Kouzine, F. et al. Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 4, 344–356.e347 (2017).
Shapiro, R. & Hachmann, J. The reaction of guanine derivatives with 1,2-dicarbonyl compounds*. Biochemistry 5, 2799–2807 (1966).
Weng, X. et al. Keth-seq for transcriptome-wide RNA structure mapping. Nat. Chem. Biol. https://doi.org/10.1038/s41589-019-0459-3 (2020).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Cramer, P. Organization and regulation of gene transcription. Nature 573, 45–54 (2019).
Paule, M. R. Transcription by RNA polymerases I and III. Nucleic Acids Res. 28, 1283–1298 (2000).
Borchert, G. M., Lanier, W. & Davidson, B. L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 13, 1097–1101 (2006).
Cer, R. Z. et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Proc. Natl Acad. Sci. USA 41, D94–D100 (2013).
Henriques, T. et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41 (2018).
Warren et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Raffaella et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 9, 234–247 (2014).
Liu, W. et al. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ. 21, 1950–1960 (2014).
Wu, T., Kamikawa, Y. F. & Donohoe, M. E. Brd4’s bromodomains mediate histone H3 acetylation and chromatin remodeling in pluripotent cells through P300 and Brg1. Cell Rep. 25, 1756–1771 (2018).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Wang, D. et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474, 390–394 (2011).
Li, W. et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520 (2013).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Arner, E. et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014 (2015).
Li, W., Notani, D. & Rosenfeld, M. G. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223 (2016).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169, 13–23 (2017).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e1816 (2018).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Cho, W.-K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018).
Chong, S. et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 (2018).
Guo, Y. E. et al. Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548 (2019).
Zhou, Z. X. et al. Mapping genomic hotspots of DNA damage by a single-strand-DNA-compatible and strand-specific ChIP-seq method. Genome Res. 23, 705–715 (2013).
Khil, P. P., Smagulova, F., Brick, K. M., Camerini-Otero, R. D. & Petukhova, G. V. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res. 22, 957–965 (2012).
Lydall, D., Nikolsky, Y., Bishop, D. K. & Weinert, T. A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383, 840–843 (1996).
Wu, T., Lyu, R., He, C. Kethoxal-assisted single-stranded DNA sequencing (KAS-seq) for capturing transcription dynamics and enhancer activity. Protoc. Exch. https://doi.org/10.21203/rs.3.pex-835/v1 (2020).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhang, Y. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 38, 576–589 (2010).
Thomas-Chollier, M. et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat. Protoc. 6, 1860–1869 (2011).
Acknowledgements
We thank all He lab members for discussion. We thank B. Harada for helpful comments on the manuscript. We thank Genomics Facility at the University of Chicago for performing high-throughput sequencing (P30 CA014599). This work was supported by US National Institutes of Health (R01 HG006827, RM1 HG008935 and P01 NS097206 to C.H.). C. H. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
All authors designed experiments and interpreted the data. T.W. performed the experiments with suggestions from Q.Y.R.L. performed the bioinformatics analysis. T.W. and C.H. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The University of Chicago has filed a patent application on KAS-seq. C.H. is a scientific founder and a member of the scientific advisory board of Accent Therapeutics, Inc., and a shareholder of Epican Genetech.
Additional information
Peer review information Lei Tang was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Characterization of the N3-kethoxal-based labeling.
a, MALDI-TOF analysis of the reaction between a 16-mer DNA oligo and N3-kethoxal. The experiment was performed in duplicates with similar results obtained. b, TLC analysis of the reaction between N3-kethoxal and deoxyguanosine (dG, left) or L-arginine (L-Arg, right) after different time intervals. The N3-kethoxal-dG results were visualized by 254 nm UV light. The N3-kethoxal-L-Arg results were visualized by ninhydrin staining. The experiment was performed in duplicates with similar results obtained. c-d, The DNA yield (c) and the A260/280 ratio (d) of gDNA isolated from N3-kethoxal-treated and control cells. P values were calculated by using two-sided unpaired Student’s t-test (n = 3 independent experiments). e, Dot blot showing biotin signals of the DNA after the biotinylation reaction in the presence or absence of N3-kethoxal or biotin-DBCO. Results from two replicates were shown for each condition. The experiment was performed in duplicates with similar results obtained. f, Agarose gel image showing the profile of libraries constructed by using input and enriched DNA samples made in the presence or absence of N3-kethoxal or biotin-DBCO. Results from two replicates were shown for each condition. The experiment was performed in duplicates with similar results obtained.
Extended Data Fig. 2 KAS-seq validation and an overview of the KAS-seq profile.
a, Fingerprint plot of KAS-seq libraries and the corresponding inputs in HEK293T cells. b, Pearson correlation scatterplot between two independent KAS-seq replicates (r = 0.99) in HEK293T cells (n = 287,970 10 Kb bins in the hg19 genome). c, Peak overlaps between two independent KAS-seq replicates in HEK293T cells. The p value was calculated using two-sided Fisher’s exact test. d, Reads distributions of KAS-seq (left) and Pol II ChIP-seq (right) signals respect to different GC fractions. e, Heatmap showing reads distribution of two independent KAS-seq replicates at gene-coding regions in mESCs. f, The distribution of KAS-seq signals, ATAC-seq signals, and selected histone modifications at gene-coding regions in HEK293T cells. g, Heatmap showing the reads distribution of two KMnO4/S1 footprinting replicates (activated mouse B cells) at gene-coding regions.
Extended Data Fig. 3 KAS-seq using low input cells and mouse liver.
KAS-seq signal distribution at gene-coding regions revealed by using different numbers of HEK293T cells (n = 26,910 genes). b, Profiles of KAS-seq data at gene-coding regions using different numbers of HEK293T cells. c, Fingerprint plot of low-input KAS-seq libraries. d. Numbers of KAS-seq peaks detected by using different amounts of HEK293T cells. e, Heatmap showing reads distribution of two independent KAS-seq replicates at gene-coding regions generated by using livers from two mice. 1 M: 1 million; 10 K: 10 thousand; 5 K: 5 thousand; 1 K: 1 thousand.
Extended Data Fig. 4 Correlation between KAS-seq signals, gene expression levels, Pol II dynamics, and gene transcription states.
a, Venn diagram showing the overlap between KAS-seq peaks and Pol II ChIP-seq peaks at promotor in mESCs. The p value was calculated using two-sided Fisher’s exact test. b, Pearson correlation scatterplot (n = 24,359 genes) between KAS-seq and Pol II ChIP-seq at gene bodies in mESCs. The r value was calculated as two-tailed probability. c, Genes were grouped according to different expression levels based on RNA-seq. 10–90 percentile of data points are shown, with the center line showing the median, and the box limits showing the upper and lower quartiles. d, Metagene profile of KAS-seq signals at gene-coding regions under control, DRB treatment, and triptolide treatment conditions. e, a snapshot of KAS-seq profiles from UCSC Genome Browser under control, DRB treatment, and triptolide treatment conditions. f, Heatmaps showing KAS-seq, Pol II ChIP-seq, and GRO-seq signals on genes with four different transcription states defined by using KAS-seq.
Extended Data Fig. 5 KAS-seq shows no significant length-dependent bias and yields strong signals around TES regions.
a, A snapshot from UCSC Genome Browser showing KAS-seq and Pol II ChIP-seq profiles at the native state, and KAS-seq profile at the DRB-treated state, indicating that KAS-seq signals around TES are derived from Pol II. Autoscale setting is used for all tracks. b, KAS-seq reads densities of three groups of genes with different lengths of termination signals. c, Averaged KAS-seq reads density in the entire terminal regions in the three groups of genes defined in (b). n = 660 genes for all three groups. d, Termination index for each gene was calculated as the ratio of KAS-seq reads density on TES to its downstream 2 kb region, versus reads density on the – 200 bp to +400 bp region around TSS. e, The distribution of termination index for all genes in KAS-seq, GRO-seq, and Pol II ChIP-seq (n = 29,160 genes). For c and e, 10 - 90 percentile of data points are shown, with the center line showing the median, and the box limits showing the upper and lower quartiles. P values were calculated using two-sided unpaired Student’s t-test.
Extended Data Fig. 6 KAS-seq detects Pol I and Pol III-mediated transcription events, as well as other non-B form DNA structures and telomeric DNA regions.
a-c, Snapshots of KAS-seq signals at selected small RNA, tRNA, and rRNA loci in HEK293T cells under native, DRB treatment, and triptolide treatment conditions. d, A summary of different types of non-B form DNA structures and the number of KAS-seq peaks (under triptolide-treatment condition) detected at each type of predicted non-B form DNA regions. e, Snapshots from UCSC genome browser showing examples of KAS-seq signals under native, DRB, and triptolide-treatment conditions at different non-B form DNA regions and telomeric DNA regions. f, Enrichment of KAS-seq signals at different non-B form DNA and telomeric DNA regions showed in (d). n = 715 regions for hairpin, n = 1,643 regions for cruciform, n = 730 regions for H-DNA, n = 356 regions for quadruplex, n = 256 regions for Z-DNA, n = 29 regions for telomere.
Extended Data Fig. 7 Features of ssDNA-containing enhancers in mESCs.
a, All ATAC-seq-positive enhancers were sorted into two groups based on whether they are KAS-seq-positive or not. Heatmaps of KAS-seq, ATAC-seq, and Pol II ChIP-seq signals on these two groups are shown. b, A metagene profile showing ATAC-seq reads density on the two groups of enhancers defined in (a). c, Expression levels of genes associated with KAS-seq positive (n = 3,080 genes) and KAS-seq negative (n = 1,544 genes) enhancers defined in (a). 10 - 90 percentile of data points are shown, with the centerline showing the median, and the box limits showing the upper and lower quartiles. The p value was calculated using two-sided unpaired Student’s t-test. d, Sequence motifs enriched in ATAC-seq-positive but KAS-seq-negative enhancers from mESCs (n = 6,082 enhancers). The p values were calculated by two-sided binomial test. e, Metagene profiles of Nanog, Oct4 and Sox2 ChIP-seq read densities at denoted enhancers in mESCs. Regions within 10 kb around the enhancer centers are shown.
Extended Data Fig. 8 ssDNA-containing enhancers in HEK293T cells.
a, A group of enhancers are single-stranded in HEK293T cells. Heatmap of KAS-seq reads densities at all enhancer regions in HEK293T cells. Active and poised enhancer regions are defined by distal H3K27ac and H3K4me1 signals. Active enhancers are sub-grouped into SSEs and DSAEs. b, Distribution of H3K27ac ChIP-seq signal across all HEK293T enhancers. Super-enhancers are defined as containing exceptionally high amounts of H3K27ac. c, The number of ssDNA-containing enhancers and super-enhancers in HEK293T cells and the overlap. The p value was calculated by two-sided Fisher’s exact test. d, KAS-seq reads densities on SSEs in HEK293T cells under native and DRB-treatment conditions. e, Metagene profiles of KAS-seq, Pol II, H3K4me3, and H3K27ac ChIP-seq reads densities at denoted enhancers in HEK293T cells. Regions within 10 kb around the enhancer centers are shown. SSE: ssDNA-containing enhancers; DSAE: double-stranded active enhancers; PE: poised enhancers.
Extended Data Fig. 9 Transcription factors that preferentially bind at ssDNA-containing enhancers in HEK293T cells.
a, Metagene profiles of CTCF, YY1, SP1, SP2, MAZ, NCAPH2, KLF8, KLF9, ZNF335, ZNF341, ZBTB20, and ZBTB26 ChIP-seq reads densities at denoted enhancers in HEK293T cells. Regions within 10 kb around the enhancer centers are shown. b, Transcription factor binding motifs enriched at ssDNA-containing enhancers (n = 1,969 enhancers) in HEK293T cells with corresponding p values by using the genome as background. Only TFs with motif information in the TRANSFAC vertebrates library were analyzed. P values were calculated by two-sided binomial test. c, GREAT analysis of genes regulated by ssDNA-containing enhancers (n = 1,969 enhancers) in HEK293T cells. P values were calculated by two-sided binomial test. SSE: ssDNA-containing enhancers; DSAE: double-stranded active enhancers; PE: poised enhancers.
Extended Data Fig. 10 KAS-seq and Pol II ChIP-seq signals in response to protein condensation inhibition.
a, PCA analysis of KAS-seq data at different time points after 1,6-hexanediol treatment (n = 3,122,843 1 kb bins). b, Box plots showing normalized KAS-seq reads densities on gene bodies (from 0.5 kb downstream TSS to TES) of the genes defined as responsive to 1,6-hexanediol treatment. 10–90 percentile of data points are shown, with the center line showing the median, and the box limits showing the upper and lower quartiles. P values were calculated by using two-sided unpaired Student’s t-test. c, Heat map showing the release and movement of KAS-seq signals (left) and Pol II clusters (right) from 0 min to 60 min after 1,6-hexanediol treatment. d, Numbers of fast responsive genes defined by KAS-seq and Pol II ChIP-seq, and the overlap. The p value was calculated by two-sided Fisher’s exact test.
Supplementary information
Supplementary Information
Supplementary Protocol
Supplementary Tables 1 and 2
Supplementary Table 1: Public data used in this study; Supplementary Table 2: All high-throughput sequencing data generated in this study.
Source data
Source Data Fig. 3
Statistical Source Data
Source Data Fig. 5
Statistical Source Data
Source Data Extended Data Fig. 1
Unprocessed blots and gel images
Rights and permissions
About this article

Cite this article
Wu, T., Lyu, R., You, Q. et al. Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ. Nat Methods 17, 515–523 (2020). https://doi.org/10.1038/s41592-020-0797-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-020-0797-9
This article is cited by
-
KAS-seq profiling captures transcription dynamics during oocyte maturation
Journal of Ovarian Research (2024)
-
KARR-seq reveals cellular higher-order RNA structures and RNA–RNA interactions
Nature Biotechnology (2024)
-
Exploring transient global transcriptional changes induced by ascorbic acid revealed via atKAS-seq profiling
Functional & Integrative Genomics (2024)
-
CasKAS: direct profiling of genome-wide dCas9 and Cas9 specificity using ssDNA mapping
Genome Biology (2023)
-
The chromatin landscape of the euryarchaeon Haloferax volcanii
Genome Biology (2023)
