
Abstract
Several challenges remain in data-independent acquisition (DIA) data analysis, such as to confidently identify peptides, define integration boundaries, remove interferences, and control false discovery rates. In practice, a visual inspection of the signals is still required, which is impractical with large datasets. We present Avant-garde as a tool to refine DIA (and parallel reaction monitoring) data. Avant-garde uses a novel data-driven scoring strategy: signals are refined by learning from the dataset itself, using all measurements in all samples to achieve the best optimization. We evaluate the performance of Avant-garde using benchmark DIA datasets and show that it can determine the quantitative suitability of a peptide peak, and reach the same levels of selectivity, accuracy, and reproducibility as manual validation. Avant-garde is complementary to existing DIA analysis engines and aims to establish a strong foundation for subsequent analysis of quantitative mass spectrometry data.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
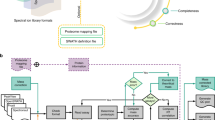


Data availability
The original mass spectra have been deposited in the public proteomics repository MassIVE and are accessible at ftp://MSV000085540@massive.ucsd.edu. Source data are provided with this paper.
Code availability
Avant-garde is an open-source software tool available as an R package and as a Skyline External tool at https://github.com/SebVaca/Avant_garde. Avant-garde can be directly downloaded from the tool Store interface within Skyline or from the Skyline tool Store at https://skyline.ms/tool-AvG.url.
References
Panchaud, A. et al. Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal. Chem. 81, 6481–6488 (2009).
Gillet, L. C. et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell Proteom. 11, O111.016717 (2012).
Egertson, J. D., MacLean, B., Johnson, R., Xuan, Y. & MacCoss, M. J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 10, 887–903 (2015).
Chapman, J. D., Goodlett, D. R. & Masselon, C. D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 33, 452–470 (2014).
Purvine, S., Eppel, J.-T., Yi, E. C. & Goodlett, D. R. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics 3, 847–850 (2003).
Silva, J. C. et al. Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 77, 2187–2200 (2005).
Silva, J. C. et al. Simultaneous qualitative and quantitative analysis of the Escherichia coli proteome: a sweet tale. Mol. Cell Proteom. 5, 589–607 (2006).
Prakash, A. et al. Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis. J. Proteome Res. 13, 5415–5430 (2014).
Geiger, T., Cox, J. & Mann, M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol. Cell Proteom. 9, 2252–2261 (2010).
Bilbao, A. et al. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 15, 964–980 (2015).
Navarro, P. et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 34, 1130–1136 (2016).
Reiter, L. et al. mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 8, 430–435 (2011).
Röst, H. L. et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 (2014).
Jaffe, J. D., Feeney, C. M., Patel, J., Lu, X. & Mani, D. R. Transitioning from targeted to comprehensive mass spectrometry using genetic algorithms. J. Am. Soc. Mass Spectrom. 27, 1745–1751 (2016).
Tsou, C.-C. et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264 (2015).
Searle, B. C. et al. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 9, 5128 (2018).
Peckner, R. et al. Specter: linear deconvolution for targeted analysis of data-independent acquisition mass spectrometry proteomics. Nat. Methods 15, 371–378 (2018).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Abelin, J. G. et al. Reduced-representation phosphosignatures measured by quantitative targeted MS capture cellular states and enable large-scale comparison of drug-induced phenotypes. Mol. Cell Proteom. 15, 1622–1641 (2016).
Rosenberger, G. et al. Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat. Methods 14, 921–927 (2017).
Röst, H. L. et al. TRIC: an automated alignment strategy for reproducible protein quantification in targeted proteomics. Nat. Methods 13, 777–783 (2016).
Ramus, C. et al. Benchmarking quantitative label-free LC-MS data processing workflows using a complex spiked proteomic standard dataset. J. Proteom. 132, 51–62 (2016).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Röst, H., Malmström, L. & Aebersold, R. A computational tool to detect and avoid redundancy in selected reaction monitoring. Mol. Cell Proteom. 11, 540–549 (2012).
Keller, A., Bader, S. L., Shteynberg, D., Hood, L. & Moritz, R. L. Automated validation of results and removal of fragment ion interferences in targeted analysis of data-independent acquisition mass spectrometry (MS) using SWATHProphet. Mol. Cell Proteom. 14, 1411–1418 (2015).
Choi, M. et al. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 (2014).
Teo, G. et al. mapDIA: preprocessing and statistical analysis of quantitative proteomics data from data independent acquisition mass spectrometry. J. Proteom. 129, 108–120 (2015).
Tsai, T.-H. et al. Selection of features with consistent profiles improves relative protein quantification in mass spectrometry experiments. Mol. Cell Proteom. 19, 944–959 (2020).
Ting, Y. S. et al. PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods 14, 903–908 (2017).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2016).
Acknowledgements
We thank N. Pythoud, J. Bons, A. Burel and C. Carapito for beta-testing the software. This work was funded by U54 HG008097 to J.D.J. This work was also supported in part by grants from the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium grants NIH/NCI U24-CA210986 and NIH/NCI U01 CA214125 (to S.A.C.) and NIH/NCI U24-CA210979 to D.R. Mani, principal computational scientist in the Proteomics Platform at the Broad Institute of MIT and Harvard.
Author information
Authors and Affiliations
Contributions
A.S.V.J. conceived the study, designed and performed experiments, collected the data, authored software, and wrote the manuscript. R.P. provided help with the data analysis of the benchmarking datasets. N.S. and B.M. adapted Skyline to facilitate the use of Avant-garde as an External tool. B.M. performed data processing and validation of the LFQBench and Extended Benchmark datasets. K.K. provided help with the statistical analysis of the benchmarking datasets. K.C.D. and A.O. carried out experiments and collected data for the P100 dataset. K.C.D., A.O. and K.E.C. beta-tested the software and provided help with the data analysis. M.J.M. and S.A.C. provided laboratory resources and guidance on the manuscript. J.D.J. provided laboratory resources, provided input on the software, provided guidance on the experimental design and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The MacCoss Lab at the University of Washington (members N.S., B.M. and M.J.M.) has a sponsored research agreement with Thermo Fisher Scientific, the manufacturer of the instrumentation used in this research. Additionally, M.J.M. is a paid consultant for Thermo Fisher Scientific. J.D.J. is employed by Inzen Therapeutics and declares that he has no conflict of interest. The remaining authors declare no competing interests.
Additional information
Peer review information Allison Doerr was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Note, Supplementary Figs. 1–15 and Supplementary Tables 1 and 2.
Supplementary Data
Supplementary Figure Source Data 1–15.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Source Data Extended Data Fig. 14
Statistical source data.
Source Data Extended Data Fig. 15
Statistical source data.
Source Data Extended Data Fig. 16
Statistical source data.
Rights and permissions
About this article

Cite this article
Vaca Jacome, A.S., Peckner, R., Shulman, N. et al. Avant-garde: an automated data-driven DIA data curation tool. Nat Methods 17, 1237–1244 (2020). https://doi.org/10.1038/s41592-020-00986-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-020-00986-4
This article is cited by
-
Deep representation features from DreamDIAXMBD improve the analysis of data-independent acquisition proteomics
Communications Biology (2021)
-
MaxDIA enables library-based and library-free data-independent acquisition proteomics
Nature Biotechnology (2021)
-
Proteomic profiling dataset of chemical perturbations in multiple biological backgrounds
Scientific Data (2021)
