
Abstract
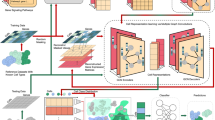
Although tremendous effort has been put into cell-type annotation, identification of previously uncharacterized cell types in heterogeneous single-cell RNA-seq data remains a challenge. Here we present MARS, a meta-learning approach for identifying and annotating known as well as new cell types. MARS overcomes the heterogeneity of cell types by transferring latent cell representations across multiple datasets. MARS uses deep learning to learn a cell embedding function as well as a set of landmarks in the cell embedding space. The method has a unique ability to discover cell types that have never been seen before and annotate experiments that are as yet unannotated. We apply MARS to a large mouse cell atlas and show its ability to accurately identify cell types, even when it has never seen them before. Further, MARS automatically generates interpretable names for new cell types by probabilistically defining a cell type in the embedding space.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The Tabula Muris Senis dataset is publicly available at https://figshare.com/projects/Tabula_Muris_Senis/64982. The Tabula Muris dataset is publicly available at https://doi.org/10.6084/m9.figshare.5829687.v8. We retrieved data from the website on 2 November 2019. We made Tabula Muris and Tabula Muris Senis datasets in h5ad format available at https://snap.stanford.edu/mars/data/tms-facs-mars.tar.gz. The Pollen dataset40 is available in the NCBI Sequence Read Archive under accession number SRP041736. Kolodziejczyk41 sequencing data are available in the ArrayExpress database under accession number E-MTAB-2600. CellBench37 and Allen Brain datasets39 are downloaded from https://doi.org/10.5281/zenodo.3357167. Originally, three brain datasets—Allen Mouse Brain (AMB), VISp and ALM—were from the Allen Institute Brain Atlas (http://celltypes.brain-map.org/rnaseq) and are available under accession number GSE115746. The CellBench 10X dataset is available under accession number GSM3618014, while CellBench CEL-Seq2dataset is from three datasets (GSM3618022, GSM3618023, GSM3618024). The project website with links to data and code can be accessed at http://snap.stanford.edu/mars/.
Code availability
MARS is written in Python using the PyTorch library. The source code is available on Github at https://github.com/snap-stanford/mars.
References
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
McKenna, A. & Gagnon, J. A. Recording development with single cell dynamic lineage tracing. Development 146, dev169730 (2019).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotech. 33, 495–502 (2015).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Han, X. et al. Mapping the mouse cell atlas by Microwell-seq. Cell 172, 1091–1107 (2018).
Schaum, N. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris: the Tabula Muris consortium. Nature 562, 367–372 (2018).
Regev, A. et al. Science forum: the Human Cell Atlas. eLife 6, e27041 (2017).
Plasschaert, L. W. et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018).
Suo, S. et al. Revealing the critical regulators of cell identity in the mouse cell atlas. Cell Rep. 25, 1436–1445 (2018).
Aevermann, B. D. et al. Cell type discovery using single-cell transcriptomics: implications for ontological representation. Hum. Mol. Genet. 27, R40–R47 (2018).
Wu, H., Kirita, Y., Donnelly, E. L. & Humphreys, B. D. Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed infibrosis. J. Am. Soc. Nephrol. 30, 23–32 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Ding, J., Condon, A. & Shah, S. P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 9, 2002 (2018).
Wang, J. et al. Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods 16, 875–878 (2019).
Amodio, M. et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods 16, 1139–1145 (2019).
Eraslan, G., Simon, L. M., Mircea, M., Mueller, N. S. & Theis, F. J. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 10, 390 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Tyssowski, K. M. et al. Different neuronal activity patterns induce different gene expression programs. Neuron 98, 530–546 (2018).
Kotliar, D. et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 8, e43803 (2019).
Pliner, H. A., Shendure, J. & Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 16, 983–986 (2019).
Xu, C. et al. Probabilistic harmonization and annotation of single-cell transcriptomics data with deep generative models. Preprint at bioRxiv https://doi.org/10.1101/532895 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Wang, T. et al. BERMUDA: a novel deep transfer learning method for single-cell RNA sequencing batch correction reveals hidden high-resolution cellular subtypes. Genome Biol. 20, 1–15 (2019).
Schmidhuber, J. Evolutionary Principles in Self-referential Learning. Diploma thesis, Technische Univ. München (1987).
Santoro, A., Bartunov, S., Botvinick, M., Wierstra, D. & Lillicrap, T. Meta-learning with memory-augmented neural networks. In Proc. International Conference on Machine Learning 33 (eds Balcan, M. F. et al.), 1842–1850 (PMLR, 2016).
Snell, J., Swersky, K. & Zemel, R. Prototypical networks for few-shot learning. Proc Adv. Neural Inform. Proc. Syst. 31 (eds Guyon, I. et al.), 4077–4087 (Curran Associates, 2017).
Finn, C., Abbeel, P. & Levine, S. Model-agnostic meta-learning for fast adaptation of deep networks. In Proc. International Conference on Machine Learning 34 (eds Precup, D. et al.) 1126–1135 (PMLR, 2017).
The Tabula Muris Consortium. A single cell transcriptomic atlas characterizes aging tissues in the mouse. Nature 583, 590–595 (2020).
Albright, J. W. & Albright, J. F. Age-associated impairment of murine natural killer activity. Proc. Natl Acad. Sci. USA 80, 6371–6375 (1983).
Nogusa, S., Ritz, B. W., Kassim, S. H., Jennings, S. R. & Gardner, E. M. Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech. Ageing Dev. 129, 223–230 (2008).
Nair, S., Fang, M. & Sigal, L. J. The natural killer cell dysfunction of aged mice is due to the bone marrow stroma and is not restored by IL-15/IL-15Rα treatment. Aging Cell 14, 180–190 (2015).
Wang, B., Zhu, J., Pierson, E., Ramazzotti, D. & Batzoglou, S. Visualization and analysis of single-cell RNA-Seq data by kernel-based similarity learning. Nat. Methods 14, 414–416 (2017).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech.: Theory Exp. 2008, P10008 (2008).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnology 36, 411–420 (2018).
Tian, L. et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 16, 479–487 (2019).
Abdelaal, T. et al. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 20, 194 (2019).
Tasic, B. et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018).
Pollen, A. A. et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotech. 32, 1053–1058 (2014).
Kolodziejczyk, A. A. et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17, 471–485 (2015).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotech. 37, 685–691 (2019).
Haniffa, M. A., Collin, M. P., Buckley, C. D. & Dazzi, F. Mesenchymal stem cells: the fibroblasts new clothes? Haematologica 94, 258–263 (2009).
Hematti, P. Mesenchymal stromal cells and fibroblasts: a case of mistaken identity? Cytotherapy 14, 516–521 (2012).
Klopfenstein, D. et al. GOATOOLS: a Python library for Gene Ontology analyses. Sci. Rep. 8, 10872 (2018).
Acknowledgements
We gratefully acknowledge the support of DARPA under nos. FA865018C7880 (ASED) and N660011924033 (MCS); ARO under nos. W911NF-16-1-0342 (MURI) and W911NF-16-1-0171 (DURIP); the National Science Foundation (NSF) under nos. OAC-1835598 (CINES), OAC-1934578 (HDR), CCF-1918940 (Expeditions) and IIS-2030477 (RAPID); the Stanford Data Science Initiative, Wu Tsai Neurosciences Institute, Chan Zuckerberg Biohub, Amazon, Boeing, Chase, Docomo, Hitachi, JD.com, NVIDIA, Dell. J.L. is a Chan Zuckerberg Biohub investigator. M.Z. is supported, in part, by NSF grant nos. IIS-2030459 and IIS-2033384, and by the Harvard Data Science Initiative. A.O.P is supported by CZ Biohub. R.B.A. is supported by CZ Biohub and grant no. NIH GM102365.
Author information
Authors and Affiliations
Contributions
M.B., M.Z. and J.L. conceived the study, designed and performed research, contributed new analytical tools, analyzed data and wrote the manuscript. M.B. also developed the software, performed experiments and developed the metrics. S.W. discussed the results and contributed to the writing. A.O.P. helped procure and interpret the datasets. S.D. and R.B.A. supervised research and contributed to the writing. J.L. supervised the research and the entire project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Lin Tang was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Brown adipose tissue embedding using PCA.
Joint low-dimensional embedding of brown adipose tissue (BAT) cell types during the life span of a mouse obtained using the PCA. We performed PCA using 100 components, corresponding to the dimensionality of low-dimensional MARS’s embeddings. Opposed to the MARS embedding space, NK cells do not change their position across different time points and are joined with the T-cells.
Extended Data Fig. 2 MARS outperforms other baselines and it is robust to embedding dimension.
Median performance of MARS and baseline methods evaluated using (a) adjusted mutual information (b) accuracy, (c) macro-F1-score, (d) macro-precision, and (e) macro-recall. For Leiden33 and Louvain34 we report adjusted mutual information and accuracy (Supplementary Note 3). Median is calculated across 21 different tissues. Error bars are standard errors estimated as a standard deviation of the mean by bootstrapping cells within tissue with n = 20 iterations. f, Median performance of MARS and K-means clustering applied in the latent space of the autoencoder at the end of the MARS pretraining. ARI stands for adjusted Rand index, F1 for macro-F1 score, and AMI for adjusted mutual information. Median is calculated across 21 different tissues. Error bars are standard errors estimated as a standard deviation of the mean by bootstrapping cells within tissue with n = 20 iterations. g, Performance of MARS when varying number of neurons in the last layer of the neural network which corresponds to the dimension of learned low-dimensional cell representation. Distribution is estimated with n = 20 runs of the method with different initial random seeds.
Extended Data Fig. 3 Cell-type level performance.
Cell-type level comparison of MARS’s F1-score with the SIMLR32 on (a) cell types that appear in only one tissue grouped by the number of differentially expressed genes in a cell type, (b) all cell types grouped by the number of differentially expressed genes in a cell type, and (c) all cell types grouped by the number of cells in a cell type. Standard errors are estimated as a standard deviation of the mean by bootstrapping cells within each tissue with n = 20 iterations. Number of differentially expressed genes is calculated using the Tabula Muris annotations by taking all genes with Benjamini-Hochberg FDR adjusted p-value<0.01 (two-tailed t-test).
Extended Data Fig. 4 Tissue-level performance.
Comparison of the MARS’s performance on individual tissues with the baseline methods. Performance is measured as adjusted Rand index score. a, Across all tissues, MARS achieves 34.3% higher area under the curve compared to the SIMLR, and 44.3% higher compared to the ScVi baseline. For each method, tissues are ranked in the decreasing order of the achieved score. b, Comparison with the second best performing method SIMLR32 on individual tissues. MARS significantly outperforms SIMLR (p = 1e − 4; two-tailed Wilcoxon signed-rank test). Tissues are ranked according to the MARS’s ARI score. Performance is measured in a single run for both methods.
Extended Data Fig. 5 Performance on other datasets.
Mean performance of MARS and four baseline methods evaluated using adjusted Rand index (Adj-Rand), F1-score (F1) and adjusted mutual information (Adj-MI) on (a) two CellBench datasets37,38 (b) Pollen40 and Kolodziejczyk clustering benchmark datasets41, and (c) three Allen Brain datasets38,39. For all metrics, higher value indicates better performance. MARS is trained in leave-one-dataset-out manner, and the held out dataset was completely unannotated.
Extended Data Fig. 6 Positive knowledge transfer on heart and mesenteric fat tissues.
Effect of the number of annotated tissues in the meta-dataset on MARS’s performance when using (a) heart tissue as unannotated experiment, and (b) mesenteric fat as unannotated experiment. Performance is measured as average adjusted Rand index across 20 runs of the method. Error bands are confidence intervals (95%) determined across 20 runs.
Extended Data Fig. 7 Embeddings after pretraining step.
UMAP visualizations of embeddings obtained with the MARS autoencoder (left), and the final MARS model (right) on (a) diaphragm tissue, and (b) liver tissue. Color indicates Tabula Muris cell type annotations. Autoencoder embeddings are obtained at the end of the MARS pretraining. Network parameters of the encoder and cluster centers from the K-means clustering are used to initialize MARS network and landmarks, respectively. MARS then learns new embeddings and new landmarks. SMS stands for skeletal muscle cell, MS for mesenchymal stem cell, HS for hepatic sinusoid, and MNKTC for mature NK T-cell.
Extended Data Fig. 8 MARS discovers cell subtypes.
a,b,UMAP visualization of MARS’s embedding of mammary gland tissue cells. Colors indicate (a) Tabula Muris cell type annotations according to Cell Ontology class, and (b) free annotations in Tabula Muris that provide additional cell type resolution. Separation of cells labeled as luminal epithelial cells into two different clusters agrees perfectly with the free annotations and separate cluster found by MARS is labeled as luminal progenitor cells. MARS also correctly assigns one basal cell misannotated as luminal epithelial cells by Cell Ontology class annotations. c,d, UMAP visualization of MARS’s embedding of subtypes of (c) basal cells of epidermis, and (d) dendritic cells. Colors indicate free annotations in Tabula Muris. We use all tissues as annotated experiments except the ones in which basal cells of epidermis or dendritic cells appear, and test the MARS ability to separate subtypes of these cell types. Clusters discovered by MARS perfectly agree with the free annotations.
Extended Data Fig. 9 Alignment of B cells.
Using MARS, B-cells in Tabula Muris data are extremely well aligned across 11 different tissues, including brown adipose tissue, diaphragm, gonodal fat, heart, kidney, limb muscle, lung, liver, mesenteric fat, subcutaneous fat, and spleen. Limb muscle is used as an unannotated tissue.
Extended Data Fig. 10 Robustness to hyperparameters.
MARS’s performance when varying (a) regularizer λ, (b) regularizer τ, and (c) number of epochs. Performance is measured as average adjusted Rand index score. Average is calculated over all tissues by including each tissue as an unannotated dataset and using other tissues as annotated experiments. Error bars are standard errors estimated as a standard deviation of the mean by bootstrapping cells within tissue with n = 20 iterations. For each value, we train MARS with all other parameters fixed.
Supplementary information
Supplementary Information
Supplementary Notes 1–4.
Rights and permissions
About this article

Cite this article
Brbić, M., Zitnik, M., Wang, S. et al. MARS: discovering novel cell types across heterogeneous single-cell experiments. Nat Methods 17, 1200–1206 (2020). https://doi.org/10.1038/s41592-020-00979-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-020-00979-3
This article is cited by
-
Computational immunogenomic approaches to predict response to cancer immunotherapies
Nature Reviews Clinical Oncology (2024)
-
Toward universal cell embeddings: integrating single-cell RNA-seq datasets across species with SATURN
Nature Methods (2024)
-
Pan-Peptide Meta Learning for T-cell receptor–antigen binding recognition
Nature Machine Intelligence (2023)
-
A scalable sparse neural network framework for rare cell type annotation of single-cell transcriptome data
Communications Biology (2023)
-
Population-level integration of single-cell datasets enables multi-scale analysis across samples
Nature Methods (2023)
