
Abstract
Single cell RNA-sequencing (scRNA-seq) technology has undergone rapid development in recent years, leading to an explosion in the number of tailored data analysis methods. However, the current lack of gold-standard benchmark datasets makes it difficult for researchers to systematically compare the performance of the many methods available. Here, we generated a realistic benchmark experiment that included single cells and admixtures of cells or RNA to create ‘pseudo cells’ from up to five distinct cancer cell lines. In total, 14 datasets were generated using both droplet and plate-based scRNA-seq protocols. We compared 3,913 combinations of data analysis methods for tasks ranging from normalization and imputation to clustering, trajectory analysis and data integration. Evaluation revealed pipelines suited to different types of data for different tasks. Our data and analysis provide a comprehensive framework for benchmarking most common scRNA-seq analysis steps.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
Raw data are available under GEO SuperSeries GSE118767. A summary of the individual accession numbers is given in Supplementary Table 1. The processed SingleCellExperiment R objects are available from https://github.com/LuyiTian/CellBench_data.
Code availability
Code used to perform the comparative analyses and generate the figures is available from https://github.com/LuyiTian/CellBench_data. The CellBench R package was developed for benchmarking single cell analysis methods and is available from https://github.com/Shians/CellBench and Bioconductor (https://www.bioconductor.org/packages/CellBench).
References
Cole, M. B. et al. Performance assessment and selection of normalization procedures for single-cell RNA-Seq. Cell Syst. 8, 315–328 (2019).
Yip, S. H., Sham, P. C. & Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief. Bioinform. https://doi.org/10.1093/bib/bby011 (2018).
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
Freytag, S., Tian, L., Lönnstedt, I., Ng, M. & Bahlo, M. Comparison of clustering tools in R for medium-sized 10x Genomics single-cell RNA-sequencing data. F1000Res. 7, 1297 (2018).
Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res. 7, 1141 (2018).
Saelens, W., Cannoodt, R., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 (2019).
Svensson, V. et al. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 14, 381–387 (2017).
Jiang, L. et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 21, 1543–1551 (2011).
Brennecke, P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1098 (2013).
Grün, D., Kester, L. & Van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Cope, L. M., Irizarry, R. A., Jaffee, H. A., Wu, Z. & Speed, T. P. A benchmark for affymetrix genechip expression measures. Bioinformatics 20, 323–331 (2004).
Sequencing Quality Control Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat. Biotechnol. 32, 903–914 (2014).
Tung, P.-Y. et al. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 7, 39921 (2017).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2018).
Huber, W. et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods 12, 115–121 (2015).
Tian, L. et al. scPipe: a flexible R/Bioconductor preprocessing pipeline for single-cell RNA-sequencing data. PLoS Comput. Biol. 14, e1006361 (2018).
Lun, A. T., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Vallejos, C. A., Marioni, J. C. & Richardson, S. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput. Biol. 11, e1004333 (2015).
Bacher, R. et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584–586 (2017).
Yip, S. H., Wang, P., Kocher, J. P. A., Sham, P. C. & Wang, J. Linnorm: improved statistical analysis for single cell RNA-seq expression data. Nucleic Acids Res. 45, e179 (2017).
Wagner, F., Yan, Y. & Yanai, I. K-nearest neighbor smoothing for high-throughput single-cell RNA-Seq data. Preprint at https://www.biorxiv.org/content/10.1101/217737v3 (2018).
Gong, W., Kwak, I.-Y., Pota, P., Koyano-Nakagawa, N. & Garry, D. J. DrImpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinformatics. 19, 220 (2018).
Huang, M. et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542 (2018).
Andrews, T. S. & Hemberg, M. False signals induced by single-cell imputation. F1000Res. 7, 1740 (2018).
Herman, J. S., Sagar & Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 15, 379–386 (2018).
Li, H. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Purdom, E. & Risso, D. clusterExperiment: Compare Clusterings for Single-Cell Sequencing. R package version 2.2.0 http://bioconductor.org/packages/3.8/bioc/html/clusterExperiment.html(2017).
Kiselev, V. Y. et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017).
Hubert, L. & Arabie, P. Comparing partitions. J. Classif. 2, 193–218 (1985).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 477 (2018).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Welch, J. D., Hartemink, A. J. & Prins, J. F. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 17, 047845 (2016).
Ji, Z. & Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 44, e117 (2016).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Haghverdi, L., Lun, A. T., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421 (2018).
Hie, B. L., Bryson, B. & Berger, B. Panoramic stitching of heterogeneous single-cell transcriptomic data. Preprint at https://www.biorxiv.org/content/10.1101/371179v1(2018).
Lin, Y. et al. scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Proc. Natl Acad. Sci. USA 116, 9775–9784 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Rohart, F., Eslami, A., Matigian, N., Bougeard, S. & Lê Cao, K.-A. MINT: a multivariate integrative method to identify reproducible molecular signatures across independent experiments and platforms. BMC Bioinformatics 18, 128 (2017).
Büttner, M., Miao, Z., Wolf, F. A., Teichmann, S. A. & Theis, F. J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49 (2019).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Holik, A. Z. et al. RNA-seq mixology: designing realistic control experiments to compare protocols and analysis methods. Nucleic Acids Res. 45, e30 (2017).
Hashimshony, T. et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77 (2016).
Muraro, M. J. et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394 (2016).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Liao, Y., Smyth, G. K. & Shi, W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
McInnes, L., Healy, J., Saul, N. & Grossberger, L. Umap: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Maechler, M., Rousseeuw, P., Struyf, A., Hubert, M. & Hornik, K. cluster: cluster analysis basics and extensions. R package version 2.0.7-1 https://cran.r-project.org/web/packages/cluster/index.html (2018).
Kang, H. M. et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018).
Scrucca, L., Fop, M., Murphy, T. B. & Raftery, A. E. mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. R J. 8, 289–317 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Kolde, R. pheatmap: Pretty Heatmaps. R package v.1.0.10 https://CRAN.R-project.org/package=pheatmap (2018).
Acknowledgements
We thank C. Weeden and M.-L. Asselin-Labat for providing the cell lines used in this study, J. Schreuder and D. Lin for assistance in conducting experiments and I. Virshup for assistance in the data integration analysis. This work was supported by funding from the National Health and Medical Research Council (NHMRC) Project Grants (No. GNT1143163 to M.E.R., No. GNT1124812 to S.H.N. and M.E.R., and No. GNT1062820 to S.H.N.), Fellowship Nos. GNT1104924 to M.E.R. and GNT1087415 to K.A.L.C., the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation (grant no. 2018-182819 to MER and no. 2018-182885 to K.A.L.C.), a Melbourne Research Scholarship to L.T., the Genomics Innovation Hub, the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS.
Author information
Authors and Affiliations
Contributions
L.T. designed, planned and performed experiments, conducted data analysis and wrote the manuscript. X.D., S.F., K.A.L.C., S.S. and A.J.A. performed data analysis and wrote the manuscript. D.A.Z., T.S.W., A.S. and J.S.J. performed experiments. S.H.N. and M.E.R. designed the study. M.E.R. supervised the analysis and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
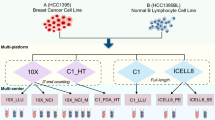
Supplementary Figure 1 Overview of the RNA mixture and cell mixture experimental designs.
(a) Ternary plot and plate layout of the RNA mixture design, which involved extracting RNA from 3 cell lines (H2228, H1975 and HCC827) in bulk, and mixing it in known proportions to get 8 mixtures that were then diluted to single cell equivalent amounts ranging from 3.75pg to 30pg. (b) Ternary plot of cell mixtures. Various 9 cell combinations were obtained using the same 3 cell lines. The number of replicates for each combination varies, as does the number of low quality samples included.
Supplementary Figure 2 Violin plots of quality control metrics for cells from each benchmarking dataset.
(a) The proportion of reads that map to introns. (b) The proportion of reads that map to exons. (c) The number of reads that map to exons. (d) The total number of counts per cell after UMI deduplication. (e) The amplification rate, which is defined by the ratio between the reads mapping to exons and the UMI counts after UMI deduplication. This measure reflects the library complexity. The sample size for each dataset is shown in Supplementary Table 1.
Supplementary Figure 3 Visualization of representative benchmarking datasets using t-SNE and UMAP and violin plot of the number of doublets.
(a) t-SNE and UMAP visualizations of 4 datasets. From left to right: 10X single cell using 3 cell lines; 10X single cell using 5 cell lines; a CEL-seq2 cell mixture and a CEL-seq2 RNA mixture. Each point represents a cell or ‘pseudo cell’ and the number of cells in each plot is indicated in the title. (b) Violin plot of doublets in each dataset, identified using Demuxlet (DBL: doublet, SNG: single cell). The number of single cells and doublets are shown on top of each violin. Doublets were excluded when calculating the performance metrics.
Supplementary Figure 4 Violin plots of silhouette widths for different normalization methods.
Silhouette widths calculated using the known biological groups after data have been normalized by different methods. The input to the silhouette width calculation is the distance between cells, which have been calculated using either (a) the gene expression matrix with the 1,000 most highly variable genes or (b) the first two PCs obtained from PCA. The sample sizes in this plots are the same as shown in Fig. 2a.
Supplementary Figure 5 Example clustering results and summary of clustering performance using ARI and the number of clusters.
(a) Examples of clustering results visualized by PCA (top) and t-SNE (bottom), with different colours representing the cluster assignments made by the selected method. The sample sizes are 340 for RNAmix_CEL-seq2, 285 for cellmix3 and 274 for sc_CEL-seq2. Coefficients obtained from linear models fitted using the ARI (b) or the number of clusters (c) as dependent variables, and experimental design, normalization methods, imputation methods and clustering methods as covariates. The coefficients measure whether particular features have positive or negative associations with the dependent variables.
Supplementary Figure 6 Coefficients from linear models used to quantify the impact different methods have on the trajectory analysis and data integration results.
Linear models were fitted using the evaluation metrics as dependent variables, with experimental design, normalization methods, imputation methods and either trajectory analysis or data integration methods as covariates. Positive coefficients indicates that a method is positively associated with the performance metrics. The evaluation metrics used as dependent variables for each plot were: (a) the correlations between calculated pseudotime and ground truth; (b) the overlap between the calculated trajectory and the known trajectory; (c) the average silhouette width of the known groups and (d) the kBET acceptance rate.
Supplementary Figure 7 Visualization of results from the trajectory analysis methods evaluated in our study.
Results for the RNAmix_Sort-seq, cellmix2 and cellmix1 analyses are shown. The dimension reduction method chosen for each method was as follows: PCA for Slingshot and TSCAN, DDR tree for Monocle2, diffusion map for DPT and locally linear embedding (LLE) for SLICER. The sample sizes are 266 for cellmix1, 268 for cellmix2 and 296 for RNAmix_Sort-seq.
Supplementary Figure 8 Additional data integration results for the single cell and RNA mixture datasets.
The kBET acceptance rate versus silhouette width coefficient for each method for the two combinations that had the highest silhouette width from the RNA mixture (a) and single cell (b) data integration analyses. The silhouette width assesses the ability of a given method to group biologically similar cells together while kBET assesses whether different batches are homogeneous after batch effect correction (scMerge_s: supervised scMerge; scMerge_us: unsupervised scMerge) (c) Additional PCA plots from the RNA mixture analysis and (d) t-SNE (perplexity = 30) plots from the single cell analysis to visualize the results of different method combinations. Cells are coloured according to protocol/batch information (t-SNE for MNNs and scanorama were based on batch corrected expression matrices). The samples sizes are n=636 and n=5,319 in panels c and d respectively.
Supplementary Figure 9 Performance of clustering on the RNA mixture datasets after data integration is applied.
Scatter plot of the ARI versus the number of clusters detected for the top performing normalization and imputation combinations. The true number of clusters for the RNA mixture experiment is 7.
Supplementary Iinformation
Supplementary Information
Supplementary Figs. 1–9
Supplementary Table 1
Summary of the benchmarking datasets generated.
Supplementary Table 2
Summary of the data characteristics and data analysis tasks that can be compared by each experimental design.
Supplementary Table 3
Summary of integrative methods used to combine data from different protocols and datasets.
Supplementary Table 4
Individual performance metrics obtained from benchmarking analysis, organized by task.
Supplementary Table 5
Run times for different analysis pipelines.
Rights and permissions
About this article

Cite this article
Tian, L., Dong, X., Freytag, S. et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods 16, 479–487 (2019). https://doi.org/10.1038/s41592-019-0425-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-019-0425-8
This article is cited by
-
A single cell RNAseq benchmark experiment embedding “controlled” cancer heterogeneity
Scientific Data (2024)
-
Domain generalization enables general cancer cell annotation in single-cell and spatial transcriptomics
Nature Communications (2024)
-
The impacts of active and self-supervised learning on efficient annotation of single-cell expression data
Nature Communications (2024)
-
The shaky foundations of simulating single-cell RNA sequencing data
Genome Biology (2023)
-
An information-theoretic approach to single cell sequencing analysis
BMC Bioinformatics (2023)
