
Abstract
A key unmet challenge in interpreting omics experiments is inferring biological meaning in the context of public functional genomics data. We developed a computational framework, Your Evidence Tailored Integration (YETI; http://yeti.princeton.edu/), which creates specialized functional interaction maps from large public datasets relevant to an individual omics experiment. Using this tailored integration, we predicted and experimentally confirmed an unexpected divergence in viral replication after seasonal or pandemic human influenza virus infection.
Similar content being viewed by others


Main
Relevant information contained in publically available functional genomics data is a potentially rich resource that can suggest follow-up experiments, complement the narrative of proposed biological models, and generate data-driven hypotheses related to an individual functional genomics experiment1,2. Currently, however, a researcher interpreting a dataset generated in the laboratory (or any specific published dataset) cannot directly leverage the global public data compendium as a source of context and insight. While these public data are far less accessible than knowledge contained in curated pathway databases or biological literature, the difficulty goes beyond accessing and analyzing the public data2. The information pertinent to an individual dataset of interest must be distilled from the massive collection of noisy and potentially irrelevant datasets, synthesized into a unified functional landscape, and used to provide biological insight.
Network-based methods provide a powerful means for mapping functional landscapes through the integration of multiple experimental modalities and data sources. The most direct approach is to construct a coexpression network, with edges capturing the degree of correlation between genes across the experimental conditions in the dataset of interest3,4,5,6 (Supplementary Fig. 1). However, because these networks are derived from gene expression measurements in a single dataset, they are susceptible to spurious correlations7,8,9. Functional networks address this challenge by identifying biologically relevant functional associations through machine-learning-based synthesis of the public data compendium9,10 (Supplementary Fig. 1). Such functional networks have been widely used to discover important new functional interactions in molecular networks, to predict the biological function of uncharacterized genes, and to recapitulate molecular response to treatments in different contexts9,10,11,12,13. A salient feature of this approach is its ability to robustly and accurately extract functional signals from very noisy and heterogeneous data collections. However, although modern functional networks can utilize the massive data compendium to identify interactions relevant to a specific tissue or biological process10,12,13, such approaches do not provide direct insight into any individual dataset. An individual experiment integrated in these collections is essentially ‘lost’ among the tens of thousands of experiments that are probabilistically integrated to generate the network.
Results
YETI networks provide an unbiased interpretation of the user dataset
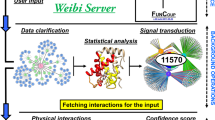
What is needed is a method to utilize the power of massive data integration while also tailoring the resulting interaction network to a specific user dataset under consideration. We therefore developed the YETI framework to provide a data-driven interpretation of any individual functional genomics experiment. There are currently no other approaches for generating functional networks from public data compendia that are specific to a given expression dataset. Previous attempts to provide information relevant to a specific dataset of interest overlaid expression data on a generic/static network, thereby identifying specific nodes or subnetworks14,15. In contrast with such overlay methods, as well as with traditional functional integration methods, YETI generates an entirely new functional network that is relevant to the dataset of interest. YETI uses the dataset of interest to guide the probabilistic Bayesian integration of the large data compendium and to generate a dataset-specific functional network (Fig. 1 and Supplementary Fig. 2). YETI relies on a library of 237 Bayesian functional networks (i.e., source networks), each capturing the biology of a particular pathway or process (Supplementary Data 2). We applied context-sensitive regularized Bayesian integration of more than 35,000 publically accessible experiments to generate specific gene–gene relationships for each source network (Methods)11,13. Together, these source networks map the entire human functional interaction landscape, although much of that landscape is not relevant to the user dataset. To automatically zoom in on only the regions of this landscape most relevant to the user dataset, YETI applies regularized linear regression to select the source functional networks with the correlation structure most closely aligned to that of the user dataset (Methods). Specifically, the dependent variable is the correlation coefficient of known functional gene–gene interactions, and the independent variables are the corresponding interaction weights of the 237 source networks (Methods). The aggregation of the selected dataset-relevant source networks results in a single, specialized YETI network of relevant functional interactions. Thus, YETI networks provide an unbiased data-driven interpretation of the user dataset and enable the generation of new hypotheses based on a combination of the researcher’s own experimental results and the relevant evidence available in public data.

YETI leverages the available public data compendium and learns global data-compendium-validated functional interactions that provide insight and predictions relevant to the user dataset.
YETI networks identify dataset-specific gene–gene interactions
In principle, the YETI framework is designed to capture both the accuracy of integrative functional network approaches and the single-experiment specificity of dataset-derived coexpression network methods (Fig. 2a). Consider a user dataset generated from breast tumor samples: an ideal integration of the public data compendium guided by this dataset would result in a network that not only correctly captures the behavior of genes in breast-cancer-relevant pathways, but also upweighs these pathways compared with interactions from pathways with no relevance to breast cancer. These properties are captured by functional accuracy, which assesses how well a network separates real functional interactions from spurious, or random, interactions, and dataset specificity, which measures whether true interactions relevant to the user dataset are upweighted by the network (Methods).

a, Schematic of trade-offs in accuracy in recovering true functional relationships (functional accuracy) and the relevance of these relationships to a specific dataset (dataset specificity). b, Comparison of dataset specificity score (DSS) and functional accuracy score (FAS) of the three network approaches for tumor datasets from the Pan-Cancer Analysis project. The different network performance assessments for the same tumor type are connected. For clarity, a representative subset of the tumor types is shown. The inset compares the performance for all Pan-Cancer tumor types (mean ± s.e.m., n = 13 Pan-Cancer tumor datasets; several s.e.m. indicators are smaller than the inset markers). c, Box plots of distributions of the DSS for generic functional networks and for YETI networks for 362 -omics datasets from GEO. In each box plot, the center line represents the median, the lower and upper hinges indicate the first and third quartiles, the upper whisker extends to the largest value less than 1.5× the interquartile range (IQR), and the lower whisker extends to the smallest value at most 1.5× the IQR. d, Box plots of distributions of the FAS for coexpression networks and for YETI networks for 362 omics datasets from GEO. Box plot elements are defined as in c. Significance was assessed by one-tailed paired t test.
We systematically compared the functional accuracy and dataset specificity of three network types: YETI networks, generic integration networks, and coexpression networks. We first used the RNA-seq datasets from The Cancer Genome Atlas (TCGA)16 (Methods). For TCGA data, just as in the hypothetical breast cancer dataset example, one can use genes that are known to be involved in the particular cancer studied in each dataset as the relevant gene set for the evaluation. We found that generic functional networks were indeed functionally accurate, but not dataset specific, whereas TCGA-dataset-derived coexpression networks were dataset specific but showed low functional accuracy (Fig. 2b). In contrast, YETI networks preserved both dataset specificity and high functional accuracy, thereby significantly outperforming both coexpression in accuracy (P = 2.4 × 10−4) and generic functional networks in specificity (P = 6.9 × 10−5) (Fig. 2b). We reached similar conclusions in a large systematic evaluation of 362 diverse disease-associated microarray datasets from Gene Expression Omnibus (GEO)17. These represent all datasets in GEO that were reported in a publication, contained more than four samples, and were associated with a specific human disease MeSH term in the annotation of the publication in PubMed. This analysis again revealed that YETI significantly outperformed generic functional networks in terms of dataset specificity (P = 1.2 × 10−8; Fig. 2c). YETI also significantly outperformed coexpression networks in accuracy (almost fivefold on average; P = 7.2 × 10−12; Fig. 2d) and dataset specificity (P = 6.7 × 10−5, one-tailed paired t test). Notably, these evaluations demonstrate YETI’s high accuracy, high dataset specificity, and broad robustness when used with either RNA-seq (TCGA) or microarray (GEO) datasets.
YETI is efficient at extracting dataset-relevant information from the data compendium
Notably, merely including the input dataset in the compendium for Bayesian integration, as opposed to YETI’s tailored approach, was not sufficient to achieve the markedly improved dataset specificity of YETI networks (Fig. 2c and Supplementary Fig. 3). In fact, the generic network shown in Fig. 2c already includes every specific user dataset used for evaluation. Furthermore, we did not observe any change in dataset specificity of the integrated generic network when we included or excluded the ‘user dataset’ from the integration process (Supplementary Fig. 4). Because each specific dataset was part of a large collection of datasets used for functional integration, the signals in the input dataset appeared to be swamped by the generic information in the massive compendium. Similarly, we found no correlation between YETI network’s performance and dataset coverage in the public compendium, which indicates that YETI is robust to irrelevant information in the public compendium and is applicable to even understudied human diseases or biological contexts (Supplementary Fig. 5). In contrast, the improvement in functional accuracy of YETI networks relative to that of coexpression networks can be attributed to YETI’s ability to control false positives in the coexpression data through the integration of this massive public compendium (Fig. 2d). Indeed, YETI eliminated the known dataset size bias of coexpression network methods (Supplementary Fig. 6) that results from erroneous but high correlations between random gene pairs, especially in small datasets7,8.
YETI leads to discoveries on the mechanism of pandemic virus infection
To illustrate the use of YETI, we applied YETI to interpret experiments from human immune cells infected with the influenza virus. There is considerable interest in understanding the differences in host–virus interactions between the historically rare pandemic influenza virus strains and the more restricted annual seasonal influenza virus strains. We applied YETI to two time-course microarray datasets from human dendritic cells, a key cell type in the immune response, after infection with either a seasonal or a pandemic wild-type influenza H1N1 virus strain. In both YETI analyses of the transcriptome response to infection, we noticed the clearly relevant “response to virus” source network (Fig. 3a,b). However, each YETI analysis also retrieved distinct source networks that were closely related to the “response to virus” network. A notable difference was the inclusion of the cell-death-related “activation of caspase activity” source network for the seasonal virus (Fig. 3a) and the essentially opposite “anti-apoptosis” source network for the pandemic virus (Fig. 3b). This difference is consistent with the finding that infection with the pandemic virus, unlike with the seasonal virus, suppresses cell death induction in dendritic cells18. A portion of one of the YETI networks (Fig. 3c) contained several nodes that have recently been implicated in programmed cell death in influenza virus infection of dendritic cells, connecting RIP–kinase pathways and apoptosis-related caspase pathway components19,20. This suggests that the YETI network can be useful for the generation of hypotheses about genes that are important in virus infection and antiviral processes.

a,b, Source networks selected from the seasonal virus dataset (a) or the pandemic virus dataset (b) that were grouped with the “response to virus” source network. Apoptosis-related source networks are highlighted in orange, and the “viral genome replication” source network is in red. c, Network neighbors of inhibitors of apoptosis (IAPs: BIRC2, BIRC3, and XIAP) and RIPK1 and RIPK3 in the YETI network from the seasonal virus dataset (edge weight threshold: 0.3). The edge colors indicate the interaction weights of the YETI network. Gold edges represent known functional interactions. d, Testing the YETI-based hypothesis by determining infectious virus titer produced in dendritic cells after infection with the seasonal (NC/99) and pandemic (Cal/09) influenza virus strains. Black diamonds represent the virus titer level for each independent experiment. Statistical comparison between seasonal and pandemic virus titer levels was performed by one-tailed t test based on n = 3 biologically independent samples 8 h and 24 h post-infection (P = 0.019 and P = 2.8 × 10−4, respectively). Data are summarized by mean and s.e.m.
Notably, another difference in source network composition between the two virus-infection YETI networks was that only the pandemic virus data identified “viral genome replication” as a relevant source network (Fig. 3a,b). This was surprising to us, as it is generally accepted that dendritic cells do not support virus replication after infection21. Motivated by the YETI detection of a virus replication signature in the pandemic infection dataset, we hypothesized that the pandemic virus, by suppressing cell death, might enable the production of new infectious virus. We experimentally tested this prediction by comparing virus replication in dendritic cells infected with the seasonal or the pandemic virus. As expected, almost no infective virus was produced after seasonal virus infection. In contrast, infection with the pandemic virus led to substantial release of new infectious virus (Fig. 3d), thus supporting the usefulness of the YETI analysis for making novel and experimentally testable inferences. These results provide an example of how researchers can use YETI to interpret and derive insights from the combination of specific datasets and public data collections that guide hypothesis generation and new experiments.
Discussion
The YETI approach leverages the biomedical knowledge embedded in the large public functional genomic data compendia to enable biomedical researchers to easily and effectively extract relevant biological signals from their omics datasets.
To facilitate broad community access to YETI, we created a user-friendly, interactive web interface at http://yeti.princeton.edu. Researchers can submit their dataset or a selected public dataset for YETI analysis to identify the biological processes reflected in the dataset-relevant source networks and interactively explore the YETI-generated network (Supplementary Fig. 3). Submitted datasets and YETI analyses remain private to the user and can be stored for the user or deleted from the server, as desired. In addition to supporting analysis of any uploaded dataset, the interface helps the user find a public dataset and corresponding dataset-specific network of interest via both text searching or relevant MeSH terms. Point-and-click access to these public YETI networks and analyses are provided for 5,300 microarray datasets from GEO17 and 1,070 RNA-seq datasets processed by Recount222. We plan to regularly update YETI’s compendium as new microarray and RNA-seq datasets become publicly available.
Thus, YETI is a complement to researchers’ biomedical knowledge and the published literature, expanding the research process with a data-driven perspective to enable new discoveries.
Methods
For a user dataset, we evaluated each network type by comparing the connectivity of genes associated with the relevant disease to random genes in the network (functional accuracy) and genes associated with other diseases (dataset specificity). In a highly accurate network, genes of the relevant disease will be more connected than random genes, and in a highly specific network, genes of the relevant disease will be much more connected than genes of other diseases. We evaluated network connectivity with curated gene sets that were independent of but relevant to the experiment. If a network focused on a specific experiment, such as breast cancer tumor samples, is highly specific to that experiment, then genes annotated to that disease should be highly connected in the network. Thus, we can assess the specificity of any network for a specific dataset on the basis of the connectivity of an independent curated relevant geneset in the network being evaluated. One source of such independent gene sets is genes annotated to MESH terms for a particular specific disease, which can be presumed to be relevant to datasets derived from clinical samples related to that disease.
Collecting and processing the public data compendium
The data compendium was compiled from multiple public databases and included gene expression profiling data, physical or genetic interaction data, transcriptional regulatory interaction data, chemical or genetic perturbation data, and microRNA target data (Supplementary Data 1). Overall, 35,300 experiments from multiple databases were collected and processed in accordance to the type of functional assay for data integration and construction of genome-wide functional networks, as described below.
Public microarray datasets (n = 980; Supplementary Data 1) that included more than 22,000 experiments were downloaded from NCBI GEO17. For each dataset, missing values were imputed using KNNImpute (K = 10)23; microarray probe identifiers were summarized to their Entrez identifiers24; and non-log-transformed values were log-transformed. For each pair of genes, the Pearson correlation (i.e., coexpression) was first computed, Fisher z-transformed, and then standardized by subtracting the mean and dividing the s.d. of the Fisher z-transformed correlations. The coexpression scores were then discretized into the following bins according to their standardized scores (i.e., z scores): ((−infinity, −1.5), [–1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, infinity)). Bins were determined empirically according to the distribution of standardized scores.
Physical and genetic interaction data were downloaded from BioGRID25, IntAct26, MINT27, and MIPS28. Interactions from BioGRID were discretized into five bins according to the number of independent supporting experiments for each interaction. Interactions from other interaction databases were discretized according to the presence or absence of the interaction. To estimate transcriptional regulatory interactions, human transcription factor (TF) binding site motifs were downloaded from JASPAR29, and for each motif, FIMO was used to identify putative regulatory regions 1 kb upstream of the gene (P < 0.001)30. We then computed TF interaction scores by taking the Pearson correlation of the TF motif profiles between two genes. Pearson correlations of the TF motif profiles were then z-transformed, standardized, and discretized with the same bin boundaries as done with the gene expression datasets.
Chemical and genetic perturbation data (C2:CGP) and microRNA target data (C3:MIR) were downloaded from the Molecular Signatures Database31. These perturbations vary widely in terms of the number of affected genes. Perturbations that result in a more restricted effect (that is, fewer genes changed) are more likely to capture more biologically specific regulatory modules, as opposed to perturbations that affect many or even most genes in the genome. To normalize for this and give more ‘weight’ to coexpression resulting from perturbations with a more restricted effect, we normalized co-occurrences by the number of genes whose expression changed in the perturbation and then aggregated them to compute the perturbation interaction score for the pair of genes. These normalized co-occurrence scores were then again discretized into bins according to their standardized z-scores: ((−infinity, −1.5), [−1.5, −0.5), [–0.5, 0.5), [0.5, 1.5), [1.5, 2.5), [2.5, 3.5), [3.5, 4.5), [4.5, infinity)). Bins were again determined empirically according to the distribution of standardized scores.
Extracting the functional training set for process-specific Bayesian network machine learning integration
Gene ontology (GO) contains experimental-evidence-based functional relationships. Known functional interactions provide a standard for distinguishing signal from noise in functional genomics data. As a result, careful extraction of known functional interactions is needed to provide a training set for effective integration of the public data compendium. In order to extract known relationships reflecting the entire biological functional landscape, we use a curated compendium of a set of GO biological processes that are testable by specific molecular experiments11. Only gene annotations based on GO experimental evidence codes (EXP, IDA, IPI, IMP, IGI, and IEP) were used to exclude potential biases from computational annotations. The resulting set of 237 GO biological process terms with at least ten gene annotations from experimental evidence codes was used to generate the training set of functional relationships (Supplementary Data 2).
To generate the training sets, we selected pairs of genes that were co-annotated to these 237 GO biological process terms after ontology propagation as highly confident, known functional interactions (positive gold-standard training set). Pairs of genes not co-annotated to any expert-curated GO term, KEGG pathway network32, PID molecular signaling or regulatory pathway33, or Biocyc metabolic pathway34 were considered as the negative gold-standard training set, except in the following conditions: (1) if two genes were annotated to two different terms with significant gene set overlap (P < 0.05), and (2) if two genes were co-annotated to a set of ‘negative’ GO terms that represented minimal functional relatedness35. Gene pairs meeting the above conditions were excluded from both positive and negative training sets.
Generation of YETI source networks
We generated functional networks presenting gene–gene relationships in each of the 237 biological processes spanning the human functional landscape (curation of 237 processes described in the section “Extracting the functional training set for process-specific Bayesian network machine learning integration” above). We applied context-sensitive, regularized Bayesian integration of the public data compendium (see “Collecting and processing the public data compendium”) for each of the 237 GO terms10,36. The resulting set of context-specific networks represent the data-driven map of the entire functional interaction landscape and are used as source networks for YETI network generation.
In detail, one regularized naive Bayes classifier was trained for each of the 237 GO term contexts. The classifier is trained based on the aforementioned positive and negative training sets (see “Extracting the functional training set for process-specific Bayesian network machine learning integration”) and using the processed public data compendium (see “Collecting and processing the public data compendium”)36. Briefly, each dataset in the compendium is modeled as a discrete probability distribution conditioned by the functional training set and the GO term context. A naive Bayes classifier without regularization assumes independence of all datasets in the compendium, and the posterior probabilities of functional gene–gene interactions are estimated with those learned parameters37. In the naive Bayes classifier, the posterior probability of functional interaction (that is, FR = 1) between genes gi and gj is
where N is the total number of datasets in the compendium, P(FR = 1) is the proportion of positives in the interaction training set, and dk(gi, gj) is the processed experimental score of gi and gj in dataset Dk.
To address the conditional-independence assumption of the naive Bayesian classifier, we used the mutual information-based regularization method that estimates the conditional dependency (that is, the amount of redundant information) and regularizes the data likelihood of the dataset10,11. Optimization of the scale parameter (that is, the strength of prior belief) in Bayesian regularization leads to greater predictive accuracy38. One can estimate this parameter by computing the amount of redundant information for each dataset10. Specifically, the estimated redundant information Uk of dataset Dk is
where I is the conditional mutual information between Di and Dk among gene pairs in the negative interaction training set, and H is the information entropy of the dataset Dk. Intuitively, an exponentially decreasing ratio is used to down-weight the likelihood function of datasets with a high amount of redundant information. Specifically, the regularized likelihood function of dataset Dk is
where P* is the regularized likelihood function for dataset Dk, dk(gi, gj) is the processed experimental score of gi and gj in dataset Dk, |Dk| is the number of discretized bins of the dataset Dk, and ns is the pseudocount hyperparameter. Pseudocount was set to 3 as described previously11. Finally, the posterior probability of any pair of genes gi and gj having a functional interaction is
where N is the total number of datasets in the compendium and P(FR = 1) is the proportion of positives in the interaction training set. This posterior probability is the interaction weight of gi and gj in the functional network. Of note, the interaction weights of the generic functional network are the average of posterior probabilities over all 237 GO term functional networks11. Software used for public data integration and distance correlation calculation has been implemented in the open source Sleipnir library available at http://libsleipnir.bitbucket.org39.
YETI’s identification of source networks relevant to the user dataset
To generate a functional network specific to the user dataset, YETI first identifies the relevant source network that was derived from the public data compendium. Optimal selection of relevant source networks is equivalent to the classic knapsack problem in computer science and is thus NP-hard40. We approximate the optimal selection by formulating this as a Lasso regression problem41 where the dependent y variable is the correlation structure of the user dataset, and the independent x variables are the 237 source networks. Lasso performs regularization (and avoids overfitting) by selecting only a subset of these independent variables, and consequently selects an approximately optimal subset of most relevant source networks. Of note, not all gene pairs are used for regression analysis, as nonfunctional interactions may exhibit high (but spurious) correlation coefficients in the user dataset. Only gene pairs of known functional interactions are considered, and their correlation coefficients (that is, interaction weights) are used to capture the specificity of the user dataset. Source networks with similar interaction weights will be predictive and thus selected in the Lasso regression analysis.
In detail, we computed the distance correlation42 of the known functional interactions (that is, positive interaction training set) of gene gi and gene gj in the user dataset Du as
where n is the total number of experiments in dataset Du, and dit is the experimental data point value of gene gi in experiment t. Distance correlation was used over other correlation statistics because of its robustness to false negatives43. Of note, the coexpression networks used as evaluation controls were also based on distance correlations.
We then used Lasso to compute a sparse solution (that is, select a relevant subset) of source networks. Consider a functional interaction m in positive interaction training set gi and gj that consists of 237 covariates \(x_m = \left( {x_{m1},x_{m2}, \ldots ,x_{m237}} \right)^T\) and a single outcome ym. xmp is the logit-transformed posterior probability of gi and gj in the ρ source network, and ym is the logit-transformed distance correlation dCorDu(gi, gj) of the user dataset Du. Specially, we used Lasso to solve
subject to
where λ ≥ 0 is the free parameter that controls the level of sparsity. The covariance test for Lasso was used to tune this level of sparsity and select significant covariates from the Lasso solution path \(\hat \beta (\lambda )\) (ref. 44). Specifically, the Lasso solution path of 100 steps is computed by the least-angle regression (LARS) algorithm45. Note that each step has a different set of active covariates. The active covariate A at the latest significant LARS step (P < 0.01) is used to determine the optimal level of sparsity and is the dataset-relevant source networks of the YETI analysis
where λ gives the latest significant LARS step. If the active set A consists of fewer than 20 covariates, the most significant LARS step (that is, smallest P value) between the 20th and 80th steps is the optimal level of sparsity, and the corresponding active set constitutes the selection of source networks identified as relevant to the user dataset. Finally, for every gene pair (gi, gj), the third quartile of the posterior probabilities among the dataset-relevant source networks was used to generate the YETI network for the user dataset. That is,
where Du is the user dataset, and \(\rho _k\left( {i,j} \right)\) is the posterior probabilities for source network k of gene pair (gi, gj). Software used for distance correlation calculation has been implemented in the open source Sleipnir library available at http://libsleipnir.bitbucket.org39. R packages lars and covTest were used for Lasso regression and covariance significance tests44,45.
Evaluating the functional accuracy and dataset specificity of interaction networks
Medical subject headings (MeSH) annotations of human genes
To evaluate knowledge-based functional networks, an independent set of gene annotations linked to the input datasets is needed. We took advantage of the Gene2MeSH (http://www.ncibi.org/gene2mesh.html) resource from the National Center for Integrative Biomedical Informatics (NCIBI), which identifies statistically significant co-occurrence of genes and MeSH terms in PubMed articles. As such, this database is independent from other databases that we used to extract known functional interactions, and also from any user dataset that we used for evaluation. Extracting this database resulted in 509 MeSH terms under the disease category with at least ten gene annotations that covered a total of 5,762 human genes. It is important to note that no disease annotations were used in any stage of YETI network construction.
Example user datasets
To emulate a typical user dataset, we downloaded all tumor-specific RNA-seq datasets with a specific tumor MeSH term ID (n = 13) from The Cancer Genome Atlas (TCGA)16 and 362 expert-curated GEO datasets17. Level-3 processed TCGA datasets of only tumors with a corresponding MeSH term were downloaded via the Cancer Genome Browser. The 362 GEO datasets represent all GEO datasets with at least one PubMed annotation, at least one MeSH term under the disease category (see below), and consist of at least four experiments.
Annotations of user datasets with MeSH terms
To evaluate the dataset specificity of interaction networks, we systematically identified MeSH terms relevant to the dataset of interest. We annotated each TCGA dataset to the MeSH ID of the corresponding cancer types. For the GEO datasets, we took advantage of the PubMed record annotated to these datasets. Each PubMed record contained associated MeSH terms, which we then assigned to the corresponding GEO dataset.
Scores for dataset specificity and functional accuracy
To assess both the dataset specificity and the functional accuracy of interaction networks, we measured the relative connectivity of genes co-annotated to MeSH terms relevant to the user dataset. The ideal interaction network useful for analyzing a single experiment must have two properties: it should be biologically accurate and specific to the user dataset. Assessment of functional accuracy entails evaluation of whether gene connections in the network represent ‘real’ interactions, for example, by being co-annotated to a particular disease. To measure this, we used MeSH-term gene annotations for the evaluation, as this source was both independent of and relevant to our collection of example user datasets (i.e., test datasets). We determined functional accuracy by measuring the pairwise connectivity of genes that were co-annotated to the dataset-relevant MeSH terms (based on curation, as described above) normalized by the connectivity of randomly selected gene pairs. Specifically, consider a graph (or network) G = (V, E), where V is the set of vertices (or genes) and E is the set of weighted edges (or interaction weights). That is, E(i, j) is the interaction weight between gene gi and gene gj in graph G. Let the gene set of the dataset-relevant MeSH term be the vertex set \({\mathrm{S}} \subset {\mathrm{V}}\). The functional accuracy score (FAS) is
where MeSH term gene set S is the set of genes in the specific MeSH term associated with the user dataset. Note that the denominator of this score is equivalent to density in graph theory46, so intuitively the measurement normalizes the relevant connectivity by overall network density.
Although functional accuracy is important, one also needs to consider how well the network is enriched for biological relationships that are relevant to the user dataset. Intuitively, a network has higher dataset specificity if it focuses on gene relationships (biological modules) that are more relevant to that dataset. Accordingly, the dataset specificity score normalizes the pairwise connectivity of genes co-annotated to the dataset-relevant MeSH terms to the pairwise connectivity of genes co-annotated to randomly selected MeSH terms. Specifically, the dataset specificity score (DSS) is calculated as
where S is the gene set for a relevant MeSH term (as above), Rw is the set of genes annotated to MeSH term w, and the median is across all 509 MeSH terms we collected from gene2mesh (see above).
Evaluating the effect of inclusion of the user dataset as part of the data compendium
Inclusion of the user dataset as part of the data compendium is an uncomplicated attempt to generate relevant functional networks. Although this method itself is straightforward, the sheer amount of data in the compendium is likely to overwhelm any relevant information coming from this single dataset. To test this hypothesis, we constructed generic functional networks with or without a user dataset and compared their DSS (as defined above). A dataset was repeatedly selected at random from the 362 GEO datasets. For each randomly selected GEO dataset (n = 10), we re-performed the entire regularized Bayesian integration process without that dataset, generating a corresponding ‘generic without dataset’ network. These were then each compared to the network generated with that dataset included (‘generic with dataset’, which is the complete network including all 362 GEO datasets).
Visualizing the dataset-relevant source networks
In addition to the interaction network, YETI also provides an interpretable list of dataset-relevant source networks, defining processes relevant to the dataset that are thus interesting for the user to explore. To facilitate visualization-based exploration of these lists, we grouped the corresponding selected GO terms on the basis of shared gene annotations. The statistical significance (or P value) of this overlap between two dataset-relevant GO terms was used as a measurement of closeness. The nearest-neighbor chain algorithm was applied to group the terms into distinct groups. The resulting interactive visualization is available on the YETI website for any user dataset and the included public datasets.
Preparation of dendritic cells
All human subject research studies were reviewed and approved by the institutional review board (IRB) of the Icahn School of Medicine at Mount Sinai (ISMMS). Informed consent was obtained from non-anonymous donors. Monocyte-derived DCs were obtained from buffy coats from human blood donors according to a standard protocol47. Briefly, peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by Ficoll density gradient centrifugation, and CD14+ monocytes were immunomagnetically purified and then differentiated into DCs during a 5-d incubation in DC medium containing 500 U/ml human granulocyte-macrophage colony-stimulating factor (hGM-CSF; Preprotech) and 1,000 U/ml human interleukin 4 (hIL-4; Preprotech).
Virus preparation and titration
The human isolates of H1N1 influenza A viruses A/California/4/2009 (Cal/09) and A/New Caledonia/20/1999 (NC) were propagated in specific-pathogen‐free embryonated hen’s eggs (Charles River Laboratories). Infectious titers of influenza viruses were determined by standard plaque assay on Madin-Darby canine kidney (MDCK) epithelial cells. Briefly, MDCK cells were seed in six-well plates. MDCK cells were cultured in minimal essential medium (MEM; Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 units/ml of penicillin, 100 μg/ml streptomycin (Gibco, Invitrogen), and 2 mM l-glutamine (Gibco, Invitrogen). The next day, tenfold dilutions of the virus stocks were prepared in phosphate-buffered saline (PBS; Gibco, Invitrogen) containing 0.2% bovine albumin (BA; MP Biomedicals). MDCK cells were washed with PBS and virus dilutions were placed on the monolayer and incubated for 1 h at room temperature. Then, virus dilutions were removed and the overlay media was added to the cells. The overlay media contained MEM (Gibco, Invitrogen), 0.2% BA (MP Biomedicals), oxoid agar (Thermo Fisher Scientific), 2 mM l-glutamine (Invitrogen), 100 units/ml of penicillin, 100 μg/ml streptomycin (Gibco, Invitrogen), 5% NaHCO3, and 1 μg/ml of trypsin N-tosyl-l-phenylalanine chloromethyl ketone (TPCK; Sigma-Aldrich). MDCK cells containing the overlay media were incubated for 48 h at 37 °C to allow for plaque formation. Cell cultures were fixed with 4% formaldehyde (Fisher Scientific) for 10 min, overlay media was removed, and cells were stained with a 0.1% crystal violet solution for visualization and quantification of the plaques.
Infection of dendritic cells
Prior to each experiment, the infectivity of each virus preparation in DCs was measured by influenza virus nucleoprotein (NP) staining, and its titer was adjusted so that each strain infects approximately 60% of the DCs obtained from each of six anonymous donors. For infection of DCs, virus stocks were diluted in serum-free medium and added directly onto pelleted DCs at a multiplicity of infection (MOI) of 1. DCs were infected in triplicates. After infection in RPMI medium at 37 °C for 10 min, cells were centrifuged to remove the viral inoculation medium and resuspended in DC medium. At the specified time points, DCs were centrifuged and supernatants were collected and stored at –80 °C for later quantification of the infectious virus particles by plaque assay on MDCK cells as described above.
Virus infection microarray experiment and data processing
For the microarray analysis, cells were homogenized with QIAshredder microcentrifuge spin-columns (Qiagen) and RNA was isolated from cells with the Qiagen Micro RNeasy plus kit according to the manufacturer’s protocol (Qiagen). RNA quality was assayed by determination of the RNA integrity number using the 2100 Bioanalyzer (Agilent). For integrated fluidic circuit real-time PCR assays (Fluidigm), RNA was extracted with Agencourt RNAdvance Cell v2 (Beckman Coulter) and RNA quantity was measured with the Ribogreen system (Life Technologies) using a fluorimeter. RNA samples were processed and hybridized to HumanHT-12v4 Expression BeadChip (Illumina), NCBI RefSeq Release 38 (7 November 2009) and selected from GenBank, dbEST, and RefSeq. Arrays were processed at Yale’s Keck Biotechnology Resource Laboratory, and raw expression data were output by the Illumina GenomeStudio software. Microarray data are available through the GEO Database, accession number GSE55278. The data were log-transformed and median-normalized. We collapsed multiple probe IDs mapping to a single official gene symbol by keeping the probe ID with highest average expression. Differential expression was defined for each probe at each infection time point using two criteria: (1) an absolute fold change of at least 2 relative to time-matched AlaF control, and (2) a significant change in expression by LIMMA after correction for multiple hypothesis testing (FDR < 0.05)48. All of this analysis was performed using BioConductor software packages in R. Further details of the sample preparation and microarray data analysis are provided elsewhere18.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
Public data listed in Supplementary Data 1 were downloaded and processed using a custom script. Source networks listed in Supplementary Data 2 were generated using the Sleipnir C++ library (http://libsleipnir.bitbucket.org/). A custom R script that uses the R packages lars and covTest was used to select dataset-relevant source networks. All custom code used in this study is available from the corresponding author on request.
Data availability
The virus infection microarray data are available in GEO under accession GSE55278. Researchers may submit their data of interest for YETI analysis at http://yeti.princeton.edu/. Visualization and exploration of their YETI network and precomputed YETI networks are also available at http://yeti.princeton.edu. All data used in this study are available from the corresponding author on request.
References
Rung, J. & Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 14, 89–99 (2013).
Dolinski, K. & Troyanskaya, O. G. Implications of Big Data for cell biology. Mol. Biol. Cell 26, 2575–2578 (2015).
Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863–14868 (1998).
Stuart, J. M., Segal, E., Koller, D. & Kim, S. K. A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003).
De Smet, R. & Marchal, K. Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8, 717–729 (2010).
Song, L., Langfelder, P. & Horvath, S. Comparison of co-expression measures: mutual information, correlation, and model based indices. BMC Bioinformatics 13, 328 (2012).
Lee, H. K., Hsu, A. K., Sajdak, J., Qin, J. & Pavlidis, P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 14, 1085–1094 (2004).
Wren, J. D. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature–data divide. Bioinformatics 25, 1694–1701 (2009).
Lee, I., Blom, U. M., Wang, P. I., Shim, J. E. & Marcotte, E. M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 21, 1109–1121 (2011).
Huttenhower, C. et al. Exploring the human genome with functional maps. Genome Res. 19, 1093–1106 (2009).
Park, C. Y. et al. Functional knowledge transfer for high-accuracy prediction of under-studied biological processes. PLoS Comput. Biol. 9, e1002957 (2013).
Gorenshteyn, D. et al. Interactive big data resource to elucidate human immune pathways and diseases. Immunity 43, 605–614 (2015).
Greene, C. S. et al. Understanding multicellular function and disease with human tissue–specific networks. Nat. Genet. 47, 569–576 (2015).
Berger, B., Peng, J. & Singh, M. Computational solutions for omics data. Nat. Rev. Genet. 14, 333–346 (2013).
Cowen, L., Ideker, T., Raphael, B. J. & Sharan, R. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18, 551–562 (2017).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016).
Clough, E. & Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 1418, 93–110 (2016).
Hartmann, B. M. et al. Human dendritic cell response signatures distinguish 1918, pandemic, and seasonal H1N1 influenza viruses. J. Virol. 89, 10190–10205 (2015).
Nogusa, S. et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24 (2016).
Hartmann, B. M. et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat. Commun. 8, 1931 (2017).
Bender, A. et al. The distinctive features of influenza virus infection of dendritic cells. Immunobiology 198, 552–567 (1998).
Collado-Torres, L. et al. Reproducible RNA-seq analysis using Recount2. Nat. Biotechnol. 35, 319–321 (2017).
Troyanskaya, O. et al. Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525 (2001).
Maglott, D., Ostell, J., Pruitt, K. D. & Tatusova, T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 39, D52–D57 (2011).
Chatr-Aryamontri, A. et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 41, D816–D823 (2013).
Kerrien, S. et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 40, D841–D846 (2012).
Licata, L. et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 40, D857–D861 (2012).
Pagel, P. et al. The MIPS mammalian protein–protein interaction database. Bioinformatics 21, 832–834 (2005).
Portales-Casamar, E. et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 38, D105–D110 (2010).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Kotera, M., Hirakawa, M., Tokimatsu, T., Goto, S. & Kanehisa, M. The KEGG databases and tools facilitating omics analysis: latest developments involving human diseases and pharmaceuticals. Methods Mol. Biol. 802, 19–39 (2012).
Schaefer, C. F. et al. PID: the Pathway Interaction Database. Nucleic Acids Res. 37, D674–D679 (2009).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 40, D742–D753 (2012).
Myers, C. L., Barrett, D. R., Hibbs, M. A., Huttenhower, C. & Troyanskaya, O. G. Finding function: evaluation methods for functional genomic data. BMC Genomics 7, 187 (2006).
Myers, C. L. & Troyanskaya, O. G. Context-sensitive data integration and prediction of biological networks. Bioinformatics 23, 2322–2330 (2007).
Friedman, N., Geiger, D. & Goldszmidt, M. Bayesian network classifiers. Mach. Learn. 29, 131–163 (1997).
Steck, H. & Jaakkola, T. S. On the Dirichlet prior and Bayesian regularization. In Advances in Neural Information Processing Systems (eds Becker, S., Thrun, S. & Obermayer, K.) 713–720 (MIT Press, Boston, MA, 2002).
Huttenhower, C., Schroeder, M., Chikina, M. D. & Troyanskaya, O. G. The Sleipnir library for computational functional genomics. Bioinformatics 24, 1559–1561 (2008).
Brucker, P. An O(n) algorithm for quadratic knapsack problems. Oper. Res. Lett. 3, 163–166 (1984).
Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. B 58, 267–288 (1996).
Szekely, G. J. & Rizzo, M. L. Brownian distance covariance. Ann. Appl. Stat. 3, 1236–1265 (2009).
Simon, N. & Tibshirani, R. Comment on “Detecting novel associations in large data sets” by Reshef Et Al, Science Dec 16, 2011. arXiv Preprint at https://arxiv.org/abs/1401.7645 (2014).
Lockhart, R., Taylor, J., Tibshirani, R. J. & Tibshirani, R. A significance test for the Lasso. Ann. Stat. 42, 413–468 (2014).
Efron, B., Hastie, T., Johnstone, I. & Tibshirani, R. Least angle regression. Ann. Stat. 32, 407–451 (2004).
Diestel, R. Graph Theory (Springer, Berlin/Heidelberg, 2018).
Bordería, A. V., Hartmann, B. M., Fernandez-Sesma, A., Moran, T. M. & Sealfon, S. C. Antiviral-activated dendritic cells: a paracrine-induced response state. J. Immunol. 181, 6872–6881 (2008).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Acknowledgements
We thank R. Dannenfelser for help in processing the TCGA RNA-seq datasets and A. Krishnan for discussions regarding the network evaluations. We greatly appreciate all members of the Troyanskaya lab for their valuable advice and discussions. This work was supported in part by the NIH (grant NIH U19 AI117873 to S.C.S.; grant NIH R01 GM071966 to O.G.T.). O.G.T. is a senior fellow of the Genetic Networks program of the Canadian Institute for Advanced Research (CIFAR).
Author information
Authors and Affiliations
Contributions
Y.-s.L., E.Z., S.C.S., and O.G.T. conceived and designed the research. Y.-s.L. performed the computational analyses with contributions from C.Y.P. A.K.W., A.T., and Y.-s.L. developed the web interface. B.M.H., V.A.D., and I.R. performed the molecular experiments. Y.-s.L., E.Z., S.C.S., and O.G.T. wrote the manuscript with revisions from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Overview of other approaches.
(left) Coexpression networks are based entirely on the correlated expression in a specific dataset. This yields functional relationships that are highly relevant to the specific dataset but does not capture accurately biological pathway interactions. (right) A generic Bayesian integration using a global data compendium accurately identifies biological pathway interactions, but the functional relationships in this network have a low specificity to any specific dataset.
Supplementary Figure 2 YETI computational framework to construct dataset-specific functional networks.
Known functional interactions are categorized into 237 distinctive biological processes spanning the multifaceted interaction landscape of the human genome. The context-specific interaction network of each biological process is learned through Bayesian integration of the public data compendium. These 237 Bayesian functional networks (i.e. source networks) are then selected based on similar interaction patterns in the user dataset of interest.
Supplementary Figure 3 Example work-through of the YETI webserver.
The user first submits her omics dataset of interest in a simple tab-separated values (TSV) format with genes in rows and omics assays in columns. The web server then integrates the public data compendium in accordance to the latent data structure of the input dataset. The user can then easily explore the dataset-relevant source networks and the dataset-specific functional map of query genes to gain deeper insight into the omics dataset used as input.
Supplementary Figure 4 Effect of exclusion of a single dataset from generic or YETI integrations.
Distribution of the Dataset Specificity Score for including the dataset, excluding it, and using YETI are shown. The center line represents the median, the lower and upper hinges indicate the first and third quartiles, the upper whisker extends to the largest value less than 1.5 x IQR and the lower whisker extends to the smallest value at most 1.5 x IQR. 10 of the 362 GEO datasets used for evaluation in Fig. 2 were chosen at random to be excluded from or included into the generic integrations that were evaluated over the MeSH terms relevant to each dataset (See Supplemental Online Methods). YETI achieved significantly improved dataset specificity over generic integration (**p = 3.1 x 10-4, one-tailed paired t test), and including the dataset of interest in the generic integration had no effect on specificity (p = 0.87, one-tailed paired t test). N.S. = not significant.
Supplementary Figure 5 Evaluation of YETI network performance robustness to the number of directly relevant datasets in the data compendium.
Accuracy score of YETI networks from disease datasets were grouped by the number of datasets annotated to the disease, excluding the user dataset used for YETI analysis. Boxplots were drawn as in Supplementary Fig. 3. The sample sizes of boxplots from left to right are: 52, 36, 64, 35, and 249.
Supplementary Figure 6 Evaluation of vulnerability of the density of YETI networks and co-expression networks to dataset size.
(a) Network densities of co-expression networks exponentially decreased with greater dataset size. (b) Network densities of YETI networks were consistently low even across input datasets of different sizes.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6
Supplementary Data 1
Name and description of datasets included in the public data compendium
Supplementary Data 2
ID and name of the 237 source networks
Rights and permissions
About this article

Cite this article
Lee, Ys., Wong, A.K., Tadych, A. et al. Interpretation of an individual functional genomics experiment guided by massive public data. Nat Methods 15, 1049–1052 (2018). https://doi.org/10.1038/s41592-018-0218-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-018-0218-5
This article is cited by
-
Olga Troyanskaya
Nature Methods (2018)
