
Abstract
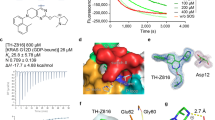
Recent progress in targeting KRASG12C has provided both insight and inspiration for targeting alternative KRAS mutants. In this study, we evaluated the mechanism of action and anti-tumor efficacy of MRTX1133, a potent, selective and non-covalent KRASG12D inhibitor. MRTX1133 demonstrated a high-affinity interaction with GDP-loaded KRASG12D with KD and IC50 values of ~0.2 pM and <2 nM, respectively, and ~700-fold selectivity for binding to KRASG12D as compared to KRASWT. MRTX1133 also demonstrated potent inhibition of activated KRASG12D based on biochemical and co-crystal structural analyses. MRTX1133 inhibited ERK1/2 phosphorylation and cell viability in KRASG12D-mutant cell lines, with median IC50 values of ~5 nM, and demonstrated >1,000-fold selectivity compared to KRASWT cell lines. MRTX1133 exhibited dose-dependent inhibition of KRAS-mediated signal transduction and marked tumor regression (≥30%) in a subset of KRASG12D-mutant cell-line-derived and patient-derived xenograft models, including eight of 11 (73%) pancreatic ductal adenocarcinoma (PDAC) models. Pharmacological and CRISPR-based screens demonstrated that co-targeting KRASG12D with putative feedback or bypass pathways, including EGFR or PI3Kα, led to enhanced anti-tumor activity. Together, these data indicate the feasibility of selectively targeting KRAS mutants with non-covalent, high-affinity small molecules and illustrate the therapeutic susceptibility and broad dependence of KRASG12D mutation-positive tumors on mutant KRAS for tumor cell growth and survival.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
The eccDNA sequencing data and RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) with accession number GSE201412. Link to the GEO public site: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201412. The datasets generated in the current study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Code availability
The eccDNA sequencing data and RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) with accession number GSE201412. Link to the GEO public site: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201412.
References
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337 (2018).
Simanshu, D. K., Nissley, D. V. & McCormick, F. RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Hallin, J. et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71 (2020).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Li, S., Balmain, A. & Counter, C. M. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer 18, 767–777 (2018).
Hunter, J. C. et al. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res 13, 1325–1335 (2015).
Lito, P., Solomon, M., Li, L. S., Hansen, R. & Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 (2016).
Patricelli, M. P. et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 6, 316–329 (2016).
Wang, X. et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J. Med. Chem. 65, 3123–3133 (2022).
Olson, P. & Hanahan, D. Cancer. Breaching the cancer fortress. Science 324, 1400–1401 (2009).
Whatcott, C. J., Posner, R. G., Von Hoff, D. D. & Han, H. Desmoplasia and chemoresistance in pancreatic cancer. In Pancreatic Cancer and Tumor Microenvironment (eds Grippo, P. J. & Munshi, H. G.) (Trivandrum, 2012).
Janne, P. et al. KRYSTAL-1: updated safety and efficacy data with adagrasib (MRTX849) in NSCLC with KRASG12C mutation from a phase 1/2 study. https://www.mirati.com/wp-content/uploads/Janne-849-001_NSCLC-ENA-Presentation_25Oct2020_FINAL.pdf (2020).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
He, L. et al. Methods for high-throughput drug combination screening and synergy scoring. Methods Mol. Biol. 1711, 351–398 (2018).
Cunningham, D. et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 351, 337–345 (2004).
Navas, C. et al. EGF receptor signaling is essential for K-Ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22, 318–330 (2012).
Lievre, A. et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 66, 3992–3995 (2006).
The Cancer Genome Atlas Network.Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Bailey, P. et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016).
Yaeger, R. et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125–136 (2018).
Janes, M. R. et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589 (2018).
Christensen, J. Discovery and characterization of MRTX1133, a selective, non-covalent inhibitor of KRASG12D. https://mirati.com/wp-content/uploads/ENA_Oct-2021_MRTX1133_vF.pdf (2021).
Weiss, J. et al. KRYSTAL-1: adagrasib (MRTX849) as monotherapy or in combination with cetuximab in patients with colorectal cancer harboring a KRASG12C mutation. https://www.mirati.com/wp-content/uploads/KRYSTAL-1-Adagrasib-MRTX849-Monotherapy-Cetuximab-CRC-KRASG12C_final.pdf (2021).
Feldmann, G., Beaty, R., Hruban, R. H. & Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 14, 224–232 (2007).
Kanda, M. et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 142, 730–733 (2012).
Zill, O. A. et al. The landscape of actionable genomic alterations in cell-free circulating tumor DNA from 21,807 advanced cancer patients. Clin. Cancer Res. 24, 3528–3538 (2018).
Chalmers, Z. R. et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Campbell, J. D. et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 (2016).
Rieder, S., Michalski, C. W., Friess, H. & Kleeff, J. Insulin-like growth factor signaling as a therapeutic target in pancreatic cancer. Anticancer Agents Med. Chem. 11, 427–433 (2011).
Rozengurt, E., Sinnett-Smith, J. & Kisfalvi, K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin. Cancer Res. 16, 2505–2511 (2010).
Ulanet, D. B., Ludwig, D. L., Kahn, C. R. & Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc. Natl Acad. Sci. USA 107, 10791–10798 (2010).
Cespedes, M. V. et al. K-ras Asp12 mutant neither interacts with Raf, nor signals through Erk and is less tumorigenic than K-ras Val12. Carcinogenesis 27, 2190–2200 (2006).
Ihle, N. T. et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J. Natl Cancer Inst. 104, 228–239 (2012).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Clark, N. A. et al. GRcalculator: an online tool for calculating and mining dose-response data. BMC Cancer 17, 698 (2017).
Conway, T. et al. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics 28, i172–i178 (2012).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Tran, T. H. et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 12, 1176 (2021).
Acknowledgements
The authors thank Crown Bioscience and Molecular Diagnostic Services for animal study support and Flagship Biosciences for immunohistochemistry and image analysis. The authors thank InviCRO, LLC, for BLI and MRI imaging and analysis of pancreatic orthotopic models. The X-ray crystallography work described herein is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which is funded by the National Institute of General Medical Sciences from the NIH (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by an NIH-ORIP High-End Instrumentation grant (S10OD021527). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract number DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
J.H., P.O. and J.G.C. designed the study. J.L., J.M. and D.V. performed biochemical characterization. J.H., V.B., L.H., L.D.E., E.L., D.T. and N.H. performed cell-based characterization. J.H., V.B., A.C., K.B., S.W. and D.M.B. performed the animal experiments. X.W., C.S., M.M., A.B., F.S., L.R. and M.A.M. performed chemical characterization. J.D.L., R.J.G. and N.C.T. determined crystal structures. L.H., J.F. and A.P. performed RNA-seq analysis. All authors analyzed data and participated in discussions on content. J.H., P.O. and J.G.C. wrote the manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
J.H., V.B., A.C., D.M.B., L.H., L.D.E., J.L., D.V., E.L., D.T., N.H., X.W., J.D.L., R.J.G., C.R.S., N.C.T., J.R.H., L.R., M.A.M., P.O. and J.G.C. are employees and shareholders of Mirati Therapeutics, Inc. A.P. is a shareholder of Mirati Therapeutics, Inc. M.M., A.B., F.S., K.B. and S.W. were employees and shareholders of Array BioPharma. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Nabeel Bardeesy, Hana Algül and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editor: Michael Basson, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Supplemental MRTX1133 pharmacology data.
(a) MRTX1133 binding abrogates effector binding in both GDP and GTP bound KRASG12D. Left: the crystal structure of KRAS bound with GTP analog, GMPPNP, complexed with the RAS binding domain of RAF1 (PDB code: 6VJJ45,). Middle: KRASG12D•MRTX1133•GDP10 and KRASG12D•MRTX1133•GMPPCP complexes (dark green ribbon) superposed with the KRAS•GMPPNP•RAF1 complex (light green and salmon ribbons). Only the inhibitor and nucleotide analog from the KRASG12D•MRTX1133•GMPPCP complex are shown for clarity. MRTX1133- bound complexes both adopt a similar conformation regardless of nucleotide state while RAF1-competent KRAS has substantially different Switch-I and Switch-II conformations. Right: Rotation of the KRAS•RAF1 complex ~90° towards the reader and cutting away the surface of RAF1 reveals the dark green surface of the KRASG12D•MRTX1133•GMPPCP complex protruding through the RAF1 surface indicating that KRAS in this conformation does not support RAF1 binding. (b) Immunoblot protein western analyses of KRAS pathway targets in GP2D cells treated for 3 hours with MRTX1133 over a 9-point dose response. Data representative of 2 independent experiments. (c) Concentration dependent effects of MRTX1133 on ERK 1/2 phosphorylation (Thr202/Tyr204) in GP2D cells assessed by band density quantitation. Data shown as percent of DMSO-treated control normalized to 1.0 by dividing all average values by the vehicle value. Calculated percent inhibition values are shown above each dose level. (d) Immunoblot protein western analyses of KRAS pathway targets in GP2D cells treated from 1 to 72 hours with MRTX1133 at 0.1, 1, and 10 nM. Data representative of 2 independent experiments. (e) Time dependent effects of MRTX1133 on ERK 1/2 phosphorylation (Thr202/Tyr204) in GP2D cells assessed by band density quantitation. Data shown as percent of DMSO-treated control normalized to 1.0 by dividing all average values by the vehicle value. Calculated percent inhibition values are shown below each dose level and time point.
Extended Data Fig. 2 Activity of MRTX1133 in KRASG12D-mutant and non KRASG12D-mutant cell line models.
Evaluation of pERK modulation and cell viability in 2D and 3D assay formats in a panel of 25 KRASG12D and 11 non-KRASG12D cells. For pERK evaluation, an In-Cell Western blot assay was used to evaluate modulation of pERK in cells treated for 3 hours with MRTX1133 over a dose response. For cell viability evaluations, CellTiter-Glo assays were used to evaluate anti-proliferative activity of cells treated with a dose response of MRTX1133 and grown in 2D tissue culture conditions in a 3-day assay, or 3D conditions using 96-well, ULA plates in a 8-day assay. 3000 for 2D and 1000 for 3D indicates IC50 values were outside of the range of the assay.
Extended Data Fig. 3 Dose dependent plasma exposure, tolerability, pharmacodynamic modulation and efficacy of MRTX1133.
(a) MRTX1133 was administered as a single dose via intraperitoneal injection at the dose levels indicated (n = 3 mice/group). The amount of MRTX1133 detected in plasma was plotted as ng/mL concentrations over time. Data shown as mean+/− standard deviation (SD). (b) MRTX1133 was administered BID daily via intraperitoneal injection at the dose levels indicated for 28 days in mice HPAC xenograft-bearing mice (n = 5 mice per group). Data are shown as mean body weights+/− standard error of the mean (SEM). Body weights were evaluated every 3–5 days. (c) Modulation of ERK1/2 phosphorylation (Tyr202/Thr204) and MRTX1133 plasma concentration (n = 3 mice/group) with vehicle or MRTX1133 administered as a single dose to mice bearing GP2D xenografts. Data shown as mean + /− SD. (D) MRTX1133 was administered intraperitoneally to mice bearing established GP2D xenografts at 30 mg/kg twice daily (BID). Dosing was initiated when tumors were ~250 mm3 (n = 5/group). MRTX1133 was administered to mice daily until Day 27. Data are shown as mean tumor volume + /− (SEM). Tumor volumes at Day 27 were determined to be statistically significant vs vehicle control two-tailed Student’s t-test (p-value <0.05).
Extended Data Fig. 4 Anti-tumor activity of MRTX1133 in KRASG12D-mutant xenografts of colon and pancreatic cancer histology in human pre-clinical models.
(a) MRTX1133 was administered i.p. BID at 30 mg/kg to mice bearing the cell line xenograft (n = 5/group) or PDX model (n = 4/group) indicated. The % change from baseline control was calculated at Day 14 for most models. Statistical significance is shown in Supplementary Table 2. (b) Pearson correlation between in vivo efficacy and PTEN expression in cell line xenografts and PDX models. Data shown with 95% confidence interval of the estimate around the correlation line, with standard error (predicted value + /− 1.96*se), no adjustments for multiple comparisons. (c) Pearson correlation between in vivo efficacy and CDKN2A expression in cell line xenografts and PDX models. Data shown as in Panel B.
Extended Data Fig. 5 Pathology and anti-tumor activity of MRTX1133 in an orthotopic pancreatic cancer xenograft model.
(a) Bioluminescent imaging of luciferase-labeled KRASG12D-mutant AsPC-1 tumors orthotopically implanted into the pancreas of immunocompromised mice. Starting on day 7 after implant, MRTX1133 was administered i.p. BID at 30 mg/kg to mice for 28 days (n = 6/group). (b) Abdominal photon flux from mice in A. Data are shown as mean abdominal bioluminescent imaging flux+/− SEM. ‘*’ indicated tumor bioluminescence at ~Day 28 for MRTX1133 treated mice were statistically significant vs vehicle using a two-tailed Student’s t-test (p-value <0.05). (c) Tumor H&E stains from vehicle and MRTX1133-treated tumors collected and processed at the end of the study. Scale for 10X images is 300um, 20X images is 200um. (d) ERK1/2 (Thr202/Tyr204) modulation at 1 and/or 6 hours post last dose was evaluated from tumors treated with Vehicle or MRTX1133 after BID + 1 dosing schedule (n = 3/time point) or BID × 28 days (n = 4 mice per time point) in AsPC-1 pancreatic orthotopic tumors. Data are shown as mean+/− standard deviation (SD).
Extended Data Fig. 6 MRTX1133 regulates KRAS-dependent oncogenic signaling and feedback inhibitory pathways in vitro.
(a) Heatmap depicting differential expression of MAPK associated gene sets with MRTX1133 treatment in A427, LS180, SNU-1033, AsPC-1, or AGS cell lines, after 3 and 24 hours of treatment (n = 3/time point) as compared to DMSO treated cells. * = -log(FDR) > 1.3 (FDR < 0.05). (b) Heatmap depicting differential expression of hallmark tumor suppressor, pro-apoptotic, pro-survival, and cell cycle associated gene sets with MRTX1133 treatment in A427, LS180, SNU-1033, AsPC-1, or AGS cell lines, after 3 and 24 hours treatment (n = 3/time point) as compared to DMSO treated cells. * = -log(FDR) > 1.3 (FDR < 0.05).
Extended Data Fig. 7 MRTX1133-anchored CRISPR/Cas9 screens in KRASG12D-mutant cell line models of pancreatic and colon cancers.
(a) Upper plot – Genes that exhibited sgRNA dropout or enrichment in two-week, DMSO-treated HPAC cells in vitro relative to MRTX1133-treated. Log2 fold change (FC) in Day 14 MRTX1133-treated samples/Plasmid vs. log2 fold change (FC) in Day 14 DMSO-treated samples/Plasmid. Lower plot - Genes that exhibited sgRNA dropout or enrichment in two-week, DMSO-treated SUIT2 cells in vitro relative to MRTX1133-treated. Log2 fold change (FC) in Day 14 MRTX1133-treated samples/Plasmid vs. Log2 fold change (FC) in Day 14 DMSO-treated samples/Plasmid. (b) Upper plot – Genes that exhibited sgRNA dropout or enrichment in two-week, Vehicle-treated HPAC cells in vivo relative to MRTX1133-treated. Log2 fold change (FC) in Day 14 MRTX1133-treated samples/Plasmid vs. log2 fold change (FC) in Day 14 Vehicle-treated samples/Plasmid. Lower plot - Genes that exhibited sgRNA dropout or enrichment in two-week, Vehicle-treated Ls180 cells in vivo relative to MRTX1133-treated. Log2 fold change (FC) in Day 14 MRTX1133-treated samples/Plasmid vs. log2 fold change (FC) in Day 14 Vehicle-treated samples/Plasmid.
Extended Data Fig. 8 MRTX1133 combination screens.
(a) Heatmap summary of in vitro synergy scores generated by combination treatments of MRTX1133 dose response with each partner compound in a dose response, run in a 72-hour, 2D Cell Titer Glo (CTG) assay to assess viability with either single agent or in combination in 26 KRASG12D-mutant cell lines. Summary color codes indicate synergy in green, additivity in yellow, and no effect in red. Cell lines not tested with the combination are denoted in grey. (b) Overlay IC50 curves generated by combination treatments of MRTX1133 dose response with cetuximab at specific concentrations in AsPC-1, Panc 04.03, GP2D, and LS180 cells (n = 3 technical replicates/dose). Cells were treated for 24 hours followed by pERK analysis to assess modulation with either single agent or in combination. Curves in grey are combinations and shift to the left, indicating increased pERK modulation. Data shown as mean + /− standard error. (c) Overlay IC50 curves generated by combination treatments of MRTX1133 dose response with BYL-719 at specific concentrations in GP2D, LS180, and AsPC-1 cells (n = 3 technical replicates/dose). Cells were treated for 24 hours followed by pAKTS473 analysis by AlphaLISA assay platform to assess modulation with either single agent or in combination. Curves in grey are combinations and shift to the left, indicating increased pAKTS473 modulation. Data shown as mean+/− standard error.
Extended Data Fig. 9 Cetuximab combination augments MRTX1133-mediated inhibition of KRAS signaling and tumor growth.
(a) Tumor growth inhibition following treatment with MRTX1133, cetuximab, or the combination administered i.p. to mice bearing the LS180 cell line xenograft (n = 5/group). Data are shown as mean tumor volume+/− SEM. Tumor volumes for the combination group were statistically significant vs all single agent treatment groups by a two-tailed Student’s t-test (* = p-value <0.05, ** = p-value <0.001) and body weight loss was <15% for the duration of the study in all groups. (b) Tumor growth inhibition following treatment with MRTX1133, cetuximab, or the combination administered i.p. to mice bearing the SW1990, Panc 02.03, Panc 04.03, or SNU-1033 cell line xenograft (n = 5/group). Data are shown as mean tumor volume+/− SEM. Tumor volumes for the combination group were statistically significant vs all single agent treatment groups by a two-tailed Student’s t-test (* = p-value <0.05, ** = p-value <0.001). Body weight loss was <15% for the duration of the study in all groups. (c) Immunohistochemical staining for pERK from mice administered MRTX1133 BID, cetuximab, or the combination followed by tumor harvest at 6 and 24 hours post dose, or MRTX1133 dosed BID for 7 days, cetuximab at 0.25 mg/dose Q3D, or the combination administered i.p. to mice bearing the AsPC-1 cell line xenograft (n = 3/time point). Images are at 10x magnification and representative of the treatment group, scale 200um. (d) Immunohistochemical staining for pS6 from mice dosed with MRTX1133 BID, cetuximab, or the combination followed by tumor harvest at 6 and 24 hours post dose, or MRTX1133 dosed BID for 7 days, cetuximab at 0.25 mg/dose Q3D, or the combination administered i.p. to mice bearing the AsPC-1 cell line xenograft (n = 3/time point). Images are at 10x magnification and representative of the treatment group, scale 200um.. (e) Percent of tumor cells positive for pERK assessed via immunohistochemistry from mice bearing the LS180 cell line xenograft dosed with MRTX1133 BID, cetuximab, or the combination followed by tumor harvest at 6 and 24 hours post dose (n = 3/time point). Data are shown as mean+/− SD. (f) Body weights of xenograft-bearing mice on the treatment schedules shown (n = 5/group). Data are shown as mean body weights+/− SEM. In this study, GP2D cells were implanted into female NOD/SCID mice, and 2 out of the 5 animals dosed with MRTX1133 BID daily exhibited weight loss and required euthanasia. In all other studies, no appreciable body weight loss was observed with treatment. (g) Percent of tumor cells positive for Ki67 assessed via immunohistochemistry from mice bearing the AsPC-1 cell line xenograft dosed i.p. with MRTX1133, cetuximab, or the combination 24 hours after the second dose, or MRTX1133 dosed BID for 7 days, cetuximab dosed Q3D, or the combination (n = 3/time point) 24 hours after the last dose. Data are shown as mean+/− SD. Ki67 staining was statistically significant relative to vehicle using one-way ANOVA. ‘*’ indicate p-value <0.05. (h) Representative 10x immunohistochemistry images from study in G, representative of the treatment group, scale 200um.
Extended Data Fig. 10 MRTX1133 treatment regulates key KRAS-dependent oncogenic signaling and feedback inhibitory pathways in vivo.
(a) Heatmap depicting GSEA Hallmark signatures altered in the AsPC-1 xenograft model by MRTX1133 treatment, or the combination of MRTX1133 with cetuximab treatment, at 6 and 24 hours after either 3 doses (BID + 1) or BID dosing for 7 days (BID × 7) as compared to vehicle-treated AsPC-1 tumors (n = 3/time point). NES = normalized enrichment score, * = false discovery rate (FDR) < 0.25. (b) Heatmap depicting differential expression of MAPK associated gene sets in MRTX1133 treatment or MRTX1133 and cetuximab combination treatment after 6- and 24-hours treatment with either 3 doses (BID + 1) or BID dosing for 7 days (BID × 7) in AsPC-1 tumors or single dose in LS180 tumors, as compared to vehicle-treated tumors (n = 3/time point). * = -log(FDR) > 1.3 (FDR < 0.05). (c) Heatmap depicting differential expression of tumor suppressor, pro-apoptotic, pro-survival, and cell cycle associated gene sets following MRTX1133 treatment or MRTX1133 and cetuximab combination treatment after 6 and 24 hours treatment with either 3 doses (BID + 1) or BID dosing for 7 days (BID × 7) in AsPC-1 tumors or single dose in LS180 tumors, as compared to vehicle-treated tumors (n = 3/time point). ‘*’ = -log(FDR) > 1.3 (FDR < 0.05).
Supplementary information
Supplementary Information
Supplementary Figs. 1–3 with legends on each figure; Scheme 1 with methods; and raw tumor measurements for Supplementary Fig. 2
Supplementary Table 1
Antibodies used for immunohistochemistry. Supplementary Table 2: In vivo tumor model, KRAS status and percent growth inhibition or regression table. Supplementary Table 3: Antibody and reagents table. Supplementary Table 4: Cell line table. Supplementary Table 5: Data collection for crystal structure. Supplementary Table 6: CRISPR sgRNA V1 library. Supplementary Table 7: CRISPR sgRNA V2 library. Supplementary Table 8: CRISPR alignments for in vitro samples. Supplementary Table 9: CRISPR alignments for in vivo samples.
Supplementary Note 1
Synthesis scheme for MRTX1133
Source data
Source Data Fig. 1
Unprocessed western blots
Source Data Fig. 2
Individual tumor volumes for Fig. 2c,f
Source Data Fig. 3
Individual tumor volumes for Fig. 3 waterfall plot
Source Data Fig. 5
Individual tumor volumes for Fig. 5a,c
Source Data Extended Data Fig. 1
Unprocessed western blots
Source Data Extended Data Fig. 3
Individual tumor volumes for Extended Data Fig. 3d
Source Data Extended Data Fig. 4
Individual tumor volumes for Extended Data Fig. 4a
Source Data Extended Data Fig. 9
Individual tumor volumes for Extended Data Fig. 9a–c
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hallin, J., Bowcut, V., Calinisan, A. et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat Med 28, 2171–2182 (2022). https://doi.org/10.1038/s41591-022-02007-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-022-02007-7
This article is cited by
-
Targeting KRAS in cancer
Nature Medicine (2024)
-
Therapeutic developments in pancreatic cancer
Nature Reviews Gastroenterology & Hepatology (2024)
-
Another KRAS variant trapped
Nature Chemical Biology (2024)
-
From bench to bedside: current development and emerging trend of KRAS-targeted therapy
Acta Pharmacologica Sinica (2024)
-
Strain-release alkylation of Asp12 enables mutant selective targeting of K-Ras-G12D
Nature Chemical Biology (2024)
