
Abstract
On 1 August 2018, the Democratic Republic of the Congo (DRC) declared its tenth Ebola virus disease (EVD) outbreak. To aid the epidemiologic response, the Institut National de Recherche Biomédicale (INRB) implemented an end-to-end genomic surveillance system, including sequencing, bioinformatic analysis and dissemination of genomic epidemiologic results to frontline public health workers. We report 744 new genomes sampled between 27 July 2018 and 27 April 2020 generated by this surveillance effort. Together with previously available sequence data (n = 48 genomes), these data represent almost 24% of all laboratory-confirmed Ebola virus (EBOV) infections in DRC in the period analyzed. We inferred spatiotemporal transmission dynamics from the genomic data as new sequences were generated, and disseminated the results to support epidemiologic response efforts. Here we provide an overview of how this genomic surveillance system functioned, present a full phylodynamic analysis of 792 Ebola genomes from the Nord Kivu outbreak and discuss how the genomic surveillance data informed response efforts and public health decision making.
Similar content being viewed by others

Main
Since the first documented outbreak of EVD in Yambuku, DRC, in 1976, further outbreaks have occurred sporadically in that country. In June 2018, laboratory capacity for performance of whole-genome EBOV sequencing was established in the DRC at the INRB in Kinshasa. The establishment of sequencing capacity enabled genomic surveillance over the entire duration of the Nord Kivu EVD outbreak (1 August 2018 to 25 June 2020). At the time of writing, we had generated 792 full and partial genome sequences representing ~24% of laboratory-confirmed cases of EVD in the region.
Comparative analysis of pathogen genomes can support traditional epidemiologic surveillance by improving the capacity to detect and define clusters of related infections, thereby facilitating detailed investigations of spatiotemporal disease dynamics. During the 2013–2016 West African EVD outbreak, analysis of viral genomic data was used to differentiate sexual EVD transmission from standard human-to-human transmission1, and to demonstrate that large, sustained case counts were attributable to many cocirculating transmission chains of varying size2.
Genomic data were also used to detect the emergence of the A82V variant that rose to high frequency during the epidemic, perhaps due to the variant’s increased infectivity in humans3,4.
Despite its utility, genomic surveillance presents challenges for many public health agencies. Assembly and analysis of pathogen genomic data can require both advanced computational infrastructure and analysts trained in disciplines that have not historically been a part of public health, including bioinformatics, computational biology and data science5. This means that the ability of public health agencies to analyze and interpret genomic data within an epidemiologic context often lags behind laboratory capacity to perform sequencing6.
We sought to increase the utility of viral genomic data during the Nord Kivu EVD outbreak by regular generation and analysis of EBOV sequence data, releasing the results as genomic epidemiology situation reports. These reports, written in both English and French, allowed representation of interactive genomic data visualization alongside written scientific interpretations. Here we provide an overview of this end-to-end genomic surveillance system, describing sequencing intensity over the course of the Nord Kivu outbreak and patterns of data release. We then describe the broad epidemic dynamics inferred from phylogeographic analysis of all 792 publicly available EBOV genomes. Finally, we discuss how the genomic data supported public health decision making and issues that impacted the actionability of the data.
Results
Overview of the genomic surveillance system
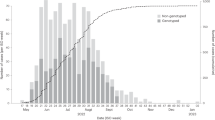
Between 27 July 2018 and 25 June 2020, clinical diagnostic specimens were collected from individuals presenting with EVD-like symptoms. A convenience sample of EBOV-positive specimens was selected for sequencing, which occurred at either a mobile laboratory in Katwa or at INRB. In total, 792 EVD genomes were sequenced: 48 of these sequences were previously published7 and 744 were analyzed here for the first time. Samples were sequenced over the full temporal span of the outbreak (Fig. 1a). While the complex geographical and political situation in eastern DRC affected sequencing intensity over time (Fig. 1a), there is minimal geographic bias. The number of sequenced cases from each health zone (the operational jurisdiction for health services in the DRC) is proportional to the total number of confirmed cases reported from that health zone (Fig. 1b).

a, Total numbers of sequenced (orange) and unsequenced (gray) laboratory-confirmed cases of EVD as reported in WHO (World Health Organization) situation reports. b, Correlation between the numbers of laboratory-confirmed and sequenced cases reported in individual health zones. c, Time lags between sample collection and release of phylogenetic analyses. In this figure, each row represents a sample. The x-axis position of a colored dot represents the date when a specific action occurred, and the color represents the action. Thus each row shows the amount of time that passed between different events for a single sequenced sample. Vertical lines represent events that occurred for a large proportion of samples; the dashed black lines represent when WHO declared that the outbreak started and ended. d,e, Kernel density estimates of lag times between sample collection and sequencing (orange), between sequencing and private release of the data (teal) and between sequencing and public release of the data (purple), before September 2019 (d) and after switching to privately released Nextstrain Narrative situation reports in September 2019 (e).
To promote open data sharing and to facilitate insights from the international scientific and public health community, genomic data were released publicly on GitHub as they were generated, accompanied by deidentified metadata (https://github.com/inrb-drc/ebola-nord-kivu). As the genomic surveillance system matured over the outbreak, the time between sequencing and data release decreased (Fig. 1c). Initially, genomic findings were communicated through haplotype maps which were manually annotated with epidemiologic information. We shared these visualizations, along with a short description of the findings, with the response team in the form of PDF files. The reports were also presented and discussed at emergency operations meetings in Goma, a city closer to the outbreak that served as a major hub for the response.
In September 2019, we transitioned from generation and manual annotation of haplotype maps to using an automated pipeline to construct divergence and temporally resolved phylogenies. We also shifted from sharing the haplotype map to writing interactive situation reports, deployed as Nextstrain Narratives8. These interactive reports allowed users to access more detailed information about the genomic data on demand, facilitating further self-guided exploration of the data if desired. The reports were available online in both English and French, and were circulated by email as PDF files that could be viewed offline. These situation reports were also presented to the public health response team at emergency operations center meetings. While the original reports contain sensitive patient information precluding public release, we have provided five deidentified reports, initially released in September and October 2019, as examples (https://nextstrain.org/community/blab/ebola-narrative-ms/).
Adopting an automated analysis pipeline increased the efficiency and scalability of analyses and reduced the average time between sequencing and private sharing of phylogenetic information (Fig. 1d,e). After adoption of the automated analysis pipeline, we shared data and analyses with the frontline response team on average within 6.6 days after sequencing (s.d. = 7.8 days). Public release of the data occurred on average 13.4 days later. The transition away from haplotype maps also enabled us to include in our analysis genomes that were less than full length and to explicitly incorporate temporal information, thereby improving the utility of these analyses for understanding disease transmission dynamics.
When circumstances were ideal, we performed diagnostic testing, sample transportation and sample preparation for sequencing in as little as 4 days, with sequencing and data analysis taking an additional 2–3 days. This timeline made it possible to deliver genomic epidemiological inferences to the response team in as few as 7 days after sample collection. However, the time period between sample collection and sequencing was typically longer. Before 1 September 2019, we sequenced and analyzed 33% (169 of 508) of samples within 30 days of collection. After September 2019 we sequenced and analyzed 48% (128 of 264 samples) within 30 days of specimen collection from patients. Notably, these proportions are conservative. Over the course of the outbreak we performed additional retrospective sequencing of archival isolates which, by definition, have longer lag times between sample collection and sequencing.
Broad-scale dynamics of EVD circulation
From phylogeographic analysis of 792 publicly available EBOV genomes collected between 27 July 2018 and 27 April 2020, we inferred broad patterns of spatial transmission over time. Previous phylogenetic analysis indicated that the Nord Kivu outbreak resulted from a single zoonotic spillover event7. We inferred that this event probably occurred in July 2018 in the Mabalako health zone (Fig. 2a), which agrees with case surveillance data7. Transmission to the nearby health zones of Beni and Mandima occurred early in the outbreak (Fig. 2a,b), with multiple introductions of EVD from Mabalako into Beni (Fig. 2a). One of these introductions, which occurred in August 2018 (95% confidence interval (CI): 15–20 August 2018), established a lineage, termed the primary outbreak clade (defined by A7312G) that became the primary circulating lineage during this outbreak (Fig. 2a). We also observed migration of viral lineages back into previously affected health zones. For example, the primary outbreak clade moved from Beni into Kalunguta around the end of August 2018 (95% CI: 16 August–12 September 2018) and was then introduced to Katwa multiple times between October 2018 and January 2019. One of the lineages circulating in Katwa then migrated back into Beni in mid-April 2019 (Fig. 2a).

a, Temporally resolved phylogenetic tree of 792 EBOV genomes, colored according to reporting health zone. The health zone of internal nodes is inferred via a discrete model, and reduced confidence is conveyed by transitioning colors to gray. b, Geographical spread of samples over four disjoint time intervals spanning the entire outbreak. Figure adapted from Nextstrain visualizations. Note that three health zones—Manguredjipa (two samples), Rwampara (four samples) and Mwenga (four samples)—are located outside of the area on the map shown here.
A secondary, sustained lineage, termed the secondary outbreak clade, resulted from an introduction from Beni into Katwa some time between August and October 2018 (Fig. 2a). This lineage later circulated in Mandima and Rwampara then migrated back into Katwa. Although smaller than the primary outbreak clade, this secondary lineage persisted throughout much of the outbreak with some genome sequences sampled as late as September 2019 clustering within this clade.
The frequent movement of viral lineages between health zones in Nord Kivu, with limited periods of local transmission after introduction, is consistent with the dynamics that sustained the West African EVD outbreak2. In that outbreak, phylogenetic analysis demonstrated that many affected regions experienced frequent independent EBOV introductions but that the subsequent transmission chains were short lived, causing on average only 75 EVD cases before dying out or moving to a new region2. Given similar apparent dynamics (Fig. 2a and Extended Data Fig. 1), we sought to quantify the frequency of EBOV introductions into health zones and the duration of local circulation after an introduction event.
In total, we detected 188 independent introduction events where the source and recipient health zones could be inferred with at least 80% confidence. Amongst these high-confidence events there were 60 distinct combinations of source health zone (where a viral lineage originated) and sink health zone (where a viral lineage moved to). Of 23 affected health zones, 11 acted only as sinks, meaning that viral lineages were introduced into that health zone but were never exported from that health zone to a different one (Extended Data Fig. 2a). The majority of introduction events into new health zones were seeded from only five source health zones: Beni, Mabalako, Katwa, Kalunguta and Mandima (Extended Data Fig. 2a,c). Each of these five health zones seeded transmission in a different health zone at least 20 separate times (Extended Data Fig. 2a).
In general, viral lineages migrated between health zones that were geographically proximal (Fig. 3a) although the geography and infrastructure of Eastern DRC means that straight-line distances may be misleading. Once introduced to a health zone, the majority of lineages circulated locally within that health zone for <8 weeks (Fig. 3d). In a minority of cases, lineages appeared to circulate locally in a health zone for much longer (Fig. 3d and Extended Data Fig. 1). It is possible that sexual transmission events from persistently infected EVD survivors artificially lengthened some of these periods, because such individuals maintain the infecting lineage over long periods of time even though that lineage is not actively circulating in the community1. On average, circulating viral lineages seeded 2.97 introduction events into new health zones, although this was highly variable (s.d. = 5.3; Fig. 3b). The length of time that a lineage circulated in a health zone was weakly, but significantly, correlated with the number of times that lineage seeded introductions into other health zones (r2 = 0.21, P < 0.001; Extended Data Fig. 2d).

a, Kernel density estimate of the inferred distance between a source and a sink health zone for 188 high-confidence events where a viral lineage moved between two health zones (HZs): 50 and 95% of movement events occurred between health zones <49 and <200 km apart, respectively. b, Kernel density estimate of the number of times a lineage was introduced into a different health zone: 50 and 95% of lineages seeded fewer than five and 25 introduction events, respectively. c, Kernel density estimate of the number of times EBOV was introduced into each health zone: 50 and 95% of health zones experienced fewer than three and eight introduction events, respectively. d, Kernel density estimate of the duration of time a lineage circulated within a single health zone: 50 and 95% of lineages circulated within a single health zone for <10 and 40 weeks, respectively.
Because these sequences represent a convenience sample of the outbreak, we performed a sensitivity analysis to evaluate the robustness of our phylogeographic inference procedure to the sampling frame. As discussed in Hall et al.9, phylogeographic analysis of sequences sampled uniformly across time and space performs similarly well to sampling demes in proportion to incidence. Thus we sampled a fraction of the full dataset to create two more equitably subsampled datasets. One dataset included three viruses sampled per health zone per month while the other included five viruses sampled per health zone per month (full and subsampled builds are available at https://nextstrain.org/community/blab/ebola-narrative-ms/). Phylogeographic analysis of these equitably subsampled datasets recapitulated the dynamics observed in analysis of the full dataset (Extended Data Fig. 3).
Case study 1: using genomic surveillance to guide vaccine allocation by detection of superspreading
Following development and testing during the West African EVD epidemic, both rVSV-ZEBOV-GP10 and Ad26-ZEBOV/MVA-BN-FILO11 vaccines were available for use during the Nord Kivu outbreak. However, given the limited supply, vaccination efforts primarily focused on contacts and contacts-of-contacts of confirmed positive cases, with preemptive vaccination available only to healthcare and frontline public health workers.
We monitored the genomic data for evidence of other settings or occupations that could be associated with high levels of secondary transmission. Consistent with previous EVD outbreaks, the data suggested that infections in clergy could contribute numerous secondary infections. For example, KAT5915 was a pastor who died of EVD in Beni. His body was transported from Beni to Butembo for burial. The funeral, which did not follow EVD safe burial protocols12, was widely attended. Exposure at the funeral led to additional cases in Beni, Butembo, Ariwara and Oicha (Extended Data Fig. 4). Three of these cases had viral genome sequences identical to KAT5915, while another seven had sequences that differed from KAT5915 by only one nucleotide (Extended Data Fig. 4). In total, 320 sequenced infections descended from this founder event.
The genomic data also suggested that secondary cases could be linked to infected motorcycle taxi drivers. For example, MAN12309 worked as a motorcycle taxi driver—including while symptomatic with EVD in December 2019. Contact tracers sought to identify exposed clients, and diagnostic specimens from clients who developed EVD were sent for sequencing. Twenty of the driver’s contacts had EBOV genome sequences identical to his, indicating that the driver was the probable source of their infection (Extended Data Fig. 5).
In response to these findings, the vaccination policy was expanded to recommend preemptive vaccination for clergy and motorcycle taxi drivers in addition to healthcare and public health workers.
Case study 2: differentiating between reinfection and relapse of a previous EVD infection
In December 2019, a male patient presented at a local health clinic with symptoms of EVD infection. In June 2019 he had become infected with EVD and sought treatment at an Ebola treatment unit in Mangina, where he recovered 14 days later. When he tested positive for EVD again in December 2019, his diagnostic specimen was sent for sequencing. Genomic analysis indicated that his December infection was genetically more similar to viral lineages that had circulated in Mabalako during June 2019 than it was to those circulating in Mabalako in December 2019. This finding prompted sequencing of his original June 2019 diagnostic specimen (Fig. 4b, annotated on the tree as MAN4194). We detected only two nucleotide differences between the driver’s June and December samples (Fig. 4c), fewer substitutions than one would expect if that viral lineage had circulated in the community for 6 months (Fig. 4a). The genomic data thus support a scenario in which the patient relapsed after recovering from his initial EVD infection, rather than having been reinfected with a different EBOV strain circulating in Mabalako in December 2019. Differentiating between these two scenarios was an important question because the patient had been vaccinated against EVD and had also received experimental monoclonal antibody treatment during his June 2019 infection. Determining whether he had relapsed or been reinfected was important for regulators seeking to understand which intervention might require further investigation. A full case report of this patient’s infections is discussed elsewhere13.

a, Root-to-tip plot showing genetic divergence of all 792 genomes as a function of their sampling date. The regression line indicates the average substitution rate across this outbreak (1.17 × 10−3 substitutions per site per year, as annotated). b, Temporally resolved phylogenetic tree showing patient’s June sample (MAN4194) and December sample (MAN12309). c, Phylogenetic tree showing nucleotide divergence from the root of this clade. The June infection (MAN4194) and December infection (MAN12309) are diverged by only two substitutions, T5587C and A6867G.
Discussion
In response to the ongoing Ebola outbreak in Nord Kivu, DRC, we implemented an end-to-end genomic surveillance system. This system included viral whole-genome sequencing, bioinformatic analysis and dissemination of genomic epidemiologic results to frontline public health workers. We used the genomic surveillance data to broadly describe epidemic dynamics. Our findings suggest that the frequent movement of viral lineages between health zones sustained the epidemic, with only a small number of lineages circulating locally within a health zone over longer periods of time. While such large-scale descriptive inferences provide important context during outbreaks, frontline public health workers also need specific, actionable pieces of information in close to real time. To meet this need, we also explored fine-scale transmission dynamics of the outbreak, monitoring for superspreading events and differentiating between relapse and reinfection events.
We began developing sequencing capability at INRB towards the end of the 2018 Equateur EVD outbreak. Our original intention was to develop the infrastructure and workforce to conduct genomic surveillance at INRB over time. However, the start of the Nord Kivu outbreak in August 2018 necessitated a faster ramp-up than we had originally intended. While the end-to-end system performed well generally, we encountered various challenges that impacted how quickly we could receive and sequence samples and thus how actionable the inferences were.
For example, sequencing capacity was initially available only in Kinshasa, roughly 2,600 km from Nord Kivu. This meant that, before sequencing, diagnostic specimens had to be transported from 11 regional diagnostic laboratories across various health zones to Beni, from Beni to Goma (~240 km) and then finally to Kinshasa (~2,400 km). Arranging specimen transport was complicated. Initially all commercial airlines flying between Goma and Kinshasa refused to carry EBOV-positive specimens. While specimen transport flights were later arranged by WHO, transport times contributed to large time lags between sample collection and sequence availability. This issue was partially mitigated by the addition of sequencing capacity at the Katwa diagnostic laboratory, starting in February 2019.
While the sequencing laboratory in Katwa improved turnaround times between sample collection and sequencing, various infrastructural, logistical and funding challenges continued to impact the speed and consistency with which we could generate sequence data. In Katwa, equipment such as gloveboxes for RNA extraction were shared between diagnostic and sequencing teams, with diagnostic teams given priority. This meant that sequencing could proceed only when diagnostic assays were complete. The high level of conflict in the region further exacerbated these delays, by limiting the number of people allowed access to the laboratory and the amount of time they could spend there. At baseline, the Katwa sequencing laboratory could not accommodate more than two scientists working simultaneously. During periods of heightened violence, such as when the Katwa Ebola Treatment Unit located next to the laboratory was destroyed by arson, access to the building was completely banned. At other times, access to the Katwa laboratory was permitted with armed escorts only, and for only 2 hours at a time, which provided insufficient time to complete steps of sequencing protocols between safe stopping points. Beyond the direct experience in Katwa, these security challenges also meant that supporting scientists were unable to travel to the outbreak area and had to provide technical support from a distance. These virtual connections were severely hampered during major Internet outages, such as the 3-week-long shut-off that occurred during the federal election in January 2019.
Finally, while funding was provided to pay for laboratory staff and space, there was no consistent funding source for purchasing of reagents. When reagents could be purchased, these were almost entirely hand-carried into the DRC by visiting international and returning Congolese scientists because traditional shipping mechanisms usually led to delays in customs during which reagents thawed and degraded. Inconsistency in the supply of sequencing reagents contributed to periods where we could not conduct sequencing despite having access to samples.
Beyond addressing these physical and logistical challenges, we believe that genomic surveillance will be more efficient and useful if it is fully integrated with traditional epidemiologic response efforts. We found that insufficient staff, limited time and the inability to travel easily to the frontline impeded communication between scientists conducting genomic surveillance and epidemiologists coordinating response efforts. This is unfortunate, because drawing inferences from multiple data sources can provide greater confidence in inferred epidemiologic dynamics and pinpoint weaknesses or erroneous findings across data streams. Integrated genomic and epidemiologic responses would also have allowed us to quantitatively evaluate how frequently genomic and surveillance epidemiological inferences aligned. A weakness of our study is that without that integration we were unable to conduct this type of evaluation. Notably, evaluation of genomic surveillance systems will be critically important to ensuring that expensive investments yield sufficient benefits, especially in low-resource settings. To support integrated surveillance systems, we will need unified databases that provide all public health responders with access to well-linked epidemiologic information, laboratory information and genomic data for cases. We also believe the system will work best if genomic and traditional epidemiologists collaborate closely in real time during outbreak response.
An additional consideration when performing genomic surveillance for outbreak response is how sampling could impact phylogeographic inference. Ideally, sampled sequences should represent the full genetic diversity of the circulating pathogen. This idealized sampling frame is often not achievable with convenience sampling during outbreaks. Therefore, as genomic surveillance becomes more common, the field would benefit from additional simulation-based work exploring how genomic epidemiologic interpretations might change as a function of sampling. Finally, phylogenetic inferences may change with the addition of more sequence data. This does not necessarily mean that the inferred dynamics are wrong; rather, one can think of the phylogeny as incomplete due to lack of data. Increasing genomic surveillance capacity such that even higher proportions of cases are sequenced will go far toward alleviating these limitations. In the meantime, genomic epidemiologists should be careful to accurately convey the meaning of the data, as well as sources of uncertainty, to surveillance epidemiologists who may be less familiar with interpreting phylogenetic trees.
Our work during the 2018–2020 EVD outbreak in Nord Kivu shows how far genomic surveillance for outbreak response has progressed. At the time, the 2013–2016 West Africa EVD epidemic was notable for its high density of sequenced cases, representing ~5% of reported EVD cases2. The vast majority of those sequences were generated by external scientists who came to West Africa, and very little sequencing capacity remained once the outbreak was declared over. Although the Nord Kivu outbreak was smaller, we sequenced close to 24% of confirmed EVD cases, with all sequencing, and now most bioinformatic analysis, occurring within the DRC. The value of building capacity within a country is demonstrated not only by our work here, but also by the sustainability of a system that can be shifted to other surveillance efforts. Indeed, using this same genomic surveillance system we are now providing much needed epidemiologic support for understanding SARS-CoV-2 epidemiology in the DRC.
Methods
Ethics statement
Diagnostic specimens were collected as part of the DRC Ministry of Health public health emergency response, and therefore consent for sample collection was waived. All preparation of samples for sequencing, genomic analysis and data analysis was performed on anonymized samples identifiable only by their laboratory or epidemiological identifier. Institutional review boards at both the United States Army Medical Research Institute of Infectious Diseases and the University of Nebraska Medical Center determined that the generation of sequencing data for public health response did not constitute research.
Sequence data generation
As described previously8, clinical diagnostic specimens were collected from individuals presenting with EVD-like symptoms. Specimens were tested for the presence of EBOV RNA using the GeneXpert Ebola Assay (Cepheid). We sequenced a subset of all EBOV-positive samples; generally, samples were sequenced if they represented an epidemiologically important case or if the case had an unusual contact history. Once samples were selected for sequencing, they were sent to either the field genomics laboratory in Katwa or INRB in Kinshasa. Samples were handled in a glovebox and RNA was extracted from the diagnostic specimen using the Viral RNA Mini kit (Qiagen). Samples were processed for sequencing using either a hybrid capture method as described previously8 or an amplicon-based method14. For hybrid capture sequencing, we used the KAPA RNA HyperPrep library preparation kit (KAPA Biosystems) with a spike-in of 20 ng of HeLa RNA (Thermo Fisher) and xGen Dual Index UMI Adapters (Integrated DNA Technologies). The libraries were enriched for EBOV using biotinylated probes (Twist Biosciences) with the TruSeq Exome Enrichment kit (Illumina). For amplicon sequencing, the Thermo Fisher first-strand synthesis system was used to reverse transcribe RNA to complementary DNA. We amplified overlapping EBOV-specific amplicons according to a primer scheme generated from PrimalSeq14 using Q5 DNA High-Fidelity Mastermix (New England Biolabs) according to the manufacturer’s specifications (primers are given in Supplementary Table 1). Amplicons were quantified with the Qubit dsDNA High Sensitivity assay on the Qubit 4.0 instrument (Life Technologies) and then diluted to <500 ng for input into library preparation. Sequencing libraries were prepared using the Illumina Nextera DNA Flex kit with IDT for Illumina Unique Dual indices. Libraries from both methods were quantified by either quantitative PCR with the KAPA Universal Library Quantification kit or Qubit with the dsDNA High Sensitivity assay, and run on an Illumina iSeq 100 or Miseq System for 2 × 150 cycles.
Bioinformatic and phylogenetic analysis
We used a custom bioinformatic pipeline to generate consensus genomes from the raw FASTQ-formatted sequencing output8,15. Deidentified metadata about the patient, diagnostic laboratory and sequence quality were paired with the consensus genome. These additional data included the laboratory identifier of the sample, the epidemiologic identifier for the patient, the patient’s symptom onset date, the sample collection date, health zone, province, laboratory that performed the diagnostic testing, sequencing date and percentage genome coverage of the sequence. Phylogenetic analysis of all consensus genomes was performed using Nextstrain16, with updated builds occurring each time new sequences were released. Alignments were verified manually in Geneious (https://www.geneious.com/).
Our specific phylogenetic analysis pipeline utilizes Augur v.6.3.0 (a component of Nextstrain), which performs a multiple sequence alignment with MAFFT v.7.402 (ref. 17), computes a maximum likelihood phylogeny using IQ-TREE v.1.6.6 (ref. 18) and temporally resolves this phylogeny using TreeTime v.0.7.2 (ref. 19). We infer the health zone at internal nodes in the tree using the discrete trait inference found in TreeTime. The resulting data are visualized using Auspice (a component of Nextstrain), which allows interactive exploration of the data.
Generation and deployment of situation reports
Following release and analysis of new sequence data, we examined the phylogenies to determine where the new sequences clustered and to investigate epidemic dynamics apparent in the genomic data. These situation reports were written in both English and French, and were shared as PDF files that could be viewed offline and as interactive reports available from a password-protected instance of nextstrain.org. Situation reports released to frontline public health workers contained sensitive patient information that necessitated private sharing. However, to illustrate what these situation reports are like, we have provided five narratives originally shared during September and October 2019, with sensitive information redacted. Links to the online interactive versions of these narratives are available at https://nextstrain.org/community/blab/ebola-narrative-ms/.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All genomic surveillance data, including consensus genomes and deidentified metadata, were released publicly over time at https://github.com/inrb-drc/ebola-nord-kivu. The exact datasets analyzed in this manuscript are available at https://github.com/blab/ebola-narrative-ms. Interactive phylogenies for the full dataset and the subsampled datasets can also be explored on Nextstrain at https://nextstrain.org/community/blab/ebola-narrative-ms/full-build, https://nextstrain.org/community/blab/ebola-narrative-ms/subsampled/3 and https://nextstrain.org/community/blab/ebola-narrative-ms/subsampled/5. Genome sequences are available on NCBI GenBank: MK007329–MK007344, MK163644–MK163675, MT778108–MT778662, MK088510 and MW797123–MW797315.
Code availability
All the code for the analyses presented in this paper, including the analysis pipeline and code for generation of figures, is available at https://github.com/blab/ebola-narrative-ms/. Nextstrain Augur and Auspice are open source, and all source code can be found at https://github.com/nextstrain/augur and https://github.com/nextstrain/auspice.
References
Mate, S. E. et al. Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 373, 2448–2454 (2015).
Dudas, G. et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 544, 309–315 (2017).
Diehl, W. E. et al. Ebola virus glycoprotein with increased infectivity dominated the 2013–2016 epidemic. Cell 167, 1088–1098 (2016).
Urbanowicz, R. A. et al. Human adaptation of Ebola virus during the West African outbreak. Cell 167, 1079–1087.e5 (2016).
Armstrong, G. L. et al. Pathogen genomics in public health. N. Engl. J. Med. 381, 2569–2580 (2019).
Black, A., MacCannell, D. R., Sibley, T. R. & Bedford, T. Ten recommendations for supporting open pathogen genomic analysis in public health. Nat. Med. 26, 832–841 (2020).
Mbala-Kingebeni, P. et al. Medical countermeasures during the 2018 Ebola virus disease outbreak in the North Kivu and Ituri provinces of the Democratic Republic of the Congo: a rapid genomic assessment. Lancet Infect. Dis. 19, 648–657 (2019).
Hadfield, J. et al. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. 15, e1008042 (2019).
Hall, M. D., Woolhouse, M. E. J. & Rambaut, A. The effects of sampling strategy on the quality of reconstruction of viral population dynamics using Bayesian skyline family coalescent methods: a simulation study. Virus Evol. 2, vew003 (2016).
Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 386, 857–866 (2015).
Milligan, I. D. et al. Safety and immunogenicity of novel adenovirus type 26—and modified vaccinia Ankara-vectored Ebola vaccines: a randomized clinical trial. JAMA 315, 1610–1623 (2016).
How to Conduct Safe and Dignified Burial of a Patient who has Died from Suspected or Confirmed Ebola or Marburg Virus Disease (World Health Organization, 2017); https://www.who.int/csr/resources/publications/ebola/safe-burial-protocol/en/
Mbala-Kingebeni, P. et al. Genomic investigation of Ebola virus transmission initiated by systemic Ebola virus disease replase. N. Engl. J. Med. (in the press).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 12, 1261–1276 (2017).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Sagulenko, P., Puller, V. & Neher, R. A. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4, vex042 (2018).
Acknowledgements
Sequencing activities were supported by the Defense Biological Product Assurance Office through a task order award to the National Strategic Research Institute (no. FA4600-12-D-9000) and Gates Foundation (no. INV-004176) awarded to C.P.P. This work was supported in part by grants from Institut National de la Santé et de la Recherche Médicale/the Ebola Task Force/REACTing, EBO-SURSY project funded by the European Union and Institut de Recherche pour le Développement (IRD). A.B. was supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1256082. P.M.-K. was awarded a PhD grant from IRD. K.G.A. is a Pew Biomedical Scholar and is supported by NIH grant nos. U01AI151812, U19AI135995 and UL1TR002550. T.B. is a Pew Biomedical Scholar and is supported by NIH grant no. R35 GM119774-01. Computational infrastructure and in-country training were supported by the Fogarty International Center (NIH/CRDF Global, no. FOGX-19-90402-1) and the Bill and Melinda Gates Foundation (no. INV-003565). The content of this article does not necessarily represent the official policy or views of the US Department of the Army, the US Department of Defense, the US Department of Health and Human Services, the US Government or the institutions or companies affiliated with the authors.
Author information
Authors and Affiliations
Contributions
E.K-.L., A.B., J.H., P.M.-K., C.B.P., M.R.W. and T.B. designed the study. E.K.-L., A.B., J.H., P.M.-K. and C.B.P. performed bioinformatic analysis and genomic epidemiologic interpretation of the data over the course of the outbreak and for this paper. D.B.M. and P.M.-K. communicated genomic analyses to frontline workers. C.B.P., B.W., M.R.W., G.P., N.D.P., E.D., M.G.P., K.G.A. and M.P. supported sequencing throughout the outbreak by both training INRB scientists and providing reagents. A.A., M.M.D., B.W., N.B., B.N. and M.A. performed the sequencing for this study. M.F., O.F., A.A.S., F.E.-A., M.-M.K., F.M.-M. and J.B. interfaced between the INRB and the frontline response. A.B., J.H., P.M.-K. and C.B.P. wrote the manuscript. G.P., E.D., A.A.S., M.P., M.R.W., S.A.-M., T.B. and J.-J.M.T. supervised this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Medicine thanks Mosoka Fallah, Yap Boum and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Jennifer Sargent was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Frequent lineage migration between health zones sustained the outbreak.
Here, the overall phylogeny (see Fig. 2 in the main text) is separated to show patterns of introduction and circulation within individual health zones for all lineages in the tree. Lineages are grouped by the health zone in which they circulated. Introductions are shown as circles at the beginning of each lineage. The color of the introduction circle indicates the donor health zone, and the x-axis position indicates the inferred timing of the introduction. While some lineages circulated in a health zone for long periods of time, most were short lived before moving into another health zone, as indicated by the relatively short branch lengths of many lineages. Visualization produced using BALTIC (github.com/evogytis/baltic/).
Extended Data Fig. 2 Patterns of transmission between health zones.
a,b, The number of introductions of EVD into a health zone positively correlates with the number of exportations out of a health zone (r2 = 0.48, p < 0.001), with most movement events occurring into and out of the same 5 health zones (Mabalako, Kalunguta, Katwa, Beni, and Mandima). State reconstructions that are less than 80% certain are excluded. c, Heatmap showing the frequency of lineage migration between all pairs of affected health zones. A migration event is counted only if the phylogeographic reconstruction for both the source and the sink health zones is at least 80% certain. d, The duration of time that a lineage circulated within a health zone is weakly correlated with the number of introduction events that a lineage seeded into other health zones (r2 = 0.21, p < 0.003).
Extended Data Fig. 3 Inferred transmission dynamics are robust to sampling.
a, Kernel density estimates for the same metrics presented in Fig. 3. This analysis used a dataset subsampled to include 3 genomes per health zone per month (total n = 323 genomes). b, Kernel density estimates for the same metrics presented in Fig. 3. This analysis used a dataset subsampled to include 5 genomes per health zone per month (total n = 433 genomes). Inferences from the subsampled datasets recapitulate the findings shown in Fig. 3, suggesting that phylogeographic inferences are robust to sampling frame.
Extended Data Fig. 4 Genomic characterization of transmission after unsafe burial of a pastor.
The horizontal axis represents nucleotide substitutions relative to the EBOV genome sequence from the pastor (KAT5915, orange). Three other samples had identical genome sequences to KAT5915. One case was from Oicha (light brown), one case was from Ariwara (neon yellow), and another was from Beni (green). Additional cases diverged by only one nucleotide were detected in Beni (green), Butembo (orange), and Kalunguta (purple).
Extended Data Fig. 5 Secondary transmission associated with infection of a motorcycle taxi driver.
The horizontal axis represents nucleotide substitutions relative to the EBOV genome sequence from the infected motorcycle taxi driver (MAN12309). Twenty other samples had identical genome sequences, as indicated in the figure by their position at 0 nucleotides diverged. Distance along the y-axis has no meaning, and only serves to separate samples for visualization. Additional sequenced cases in Mabalako were more genetically diverged from MAN12309, indicating additional propagated transmission following this event.
Supplementary information
Supplementary Information
Supplementary Table 1, with primer sequences.
Rights and permissions
About this article

Cite this article
Kinganda-Lusamaki, E., Black, A., Mukadi, D.B. et al. Integration of genomic sequencing into the response to the Ebola virus outbreak in Nord Kivu, Democratic Republic of the Congo. Nat Med 27, 710–716 (2021). https://doi.org/10.1038/s41591-021-01302-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01302-z
