
Abstract
Seasonal influenza viruses constantly change through antigenic drift and the emergence of pandemic influenza viruses through antigenic shift is unpredictable. Conventional influenza virus vaccines induce strain-specific neutralizing antibodies against the variable immunodominant globular head domain of the viral hemagglutinin protein. This necessitates frequent re-formulation of vaccines and handicaps pandemic preparedness. In this completed, observer-blind, randomized, placebo-controlled phase I trial (NCT03300050), safety and immunogenicity of chimeric hemagglutinin-based vaccines were tested in healthy, 18–39-year-old US adults. The study aimed to test the safety and ability of the vaccines to elicit broadly cross-reactive antibodies against the hemagglutinin stalk domain. Participants were enrolled into five groups to receive vaccinations with live-attenuated followed by AS03-adjuvanted inactivated vaccine (n = 20), live-attenuated followed by inactivated vaccine (n = 15), twice AS03-adjuvanted inactivated vaccine (n = 16) or placebo (n = 5, intranasal followed by intramuscular; n = 10, twice intramuscular) 3 months apart. Vaccination was found to be safe and induced a broad, strong, durable and functional immune response targeting the conserved, immunosubdominant stalk of the hemagglutinin. The results suggest that chimeric hemagglutinins have the potential to be developed as universal vaccines that protect broadly against influenza viruses.
Similar content being viewed by others

Main
Influenza virus infections are a substantial public health concern. Seasonal influenza causes between 290,000 and 650,000 deaths every year globally, according to the World Health Organization. In addition, influenza pandemics occur at irregular intervals and can claim millions of lives. The most devastating example is the H1N1 pandemic of 1918, which caused 40 million deaths according to conservative estimates1. Current seasonal influenza virus vaccines contain three or four strains of influenza virus that cover the viruses circulating in the human population. These viruses are grouped based on their phylogenetic differences into influenza A and influenza B viruses. Influenza A virus strains are further differentiated into group 1 and group 2 viruses and the H1N1 and H3N2 viruses included in the vaccine are part of these respective groups. Influenza B viruses are made up of two lineages (B/Yamagata/16/88-like and B/Victoria/2/87-like) and either one or both influenza B strains are included into seasonal vaccines each year. These vaccines work well when they are well matched to the circulating virus strains. However, the vaccine strain composition is based on a prediction and mismatches occur relatively frequently2. One of the seasonal influenza A virus subtypes, H3N2, recently split into several antigenically distinct clades that are cocirculating, which adds to the problem of vaccine mismatch3,4. Similarly, influenza B viruses from both the B/Victoria/2/87-like and the B/Yamagata/16/88-like lineages are gaining increased antigenic diversity and are therefore harder to predict and to match with a vaccine5,6. The situation is even worse for emerging pandemic viruses, since these outbreaks cannot be predicted and can emerge quickly. For new pandemic virus strains, matched vaccines need to be generated. This takes approximately 6 months, during which time the population is vulnerable7. Therefore, the development of a universal influenza virus vaccine that could protect against all influenza viruses is a major focus area for the research community.
Current influenza virus vaccines mostly target the immunodominant head domain of the viral hemagglutinin (HA) and are therefore strain specific8. These antibodies often have hemagglutination inhibition (HI) activity, which typically correlates with protection from influenza virus infection and disease9. Unfortunately, the head domain is highly plastic10,11 and the virus escapes neutralization by mutating this part of the HA, through a mechanism called antigenic drift12. The membrane-proximal stalk domain of the HA is more conserved compared with the head domain12. Monoclonal antibodies isolated from mice13 and humans14 that target this domain have been shown to broadly neutralize diverse influenza virus strains in vitro and to protect animals from influenza virus challenges. Of note, the majority of these antibodies bind to group 1, group 2 or influenza B virus HAs (Fig. 1)15,16. Antibodies that cross-react between groups have been isolated less often. Importantly, anti-stalk antibodies have recently been shown to correlate with protection from influenza virus infection in humans17. The mechanism of action of these antibodies includes direct virus neutralization, inhibition of HA activation, inhibition of egress, inhibition of the neuraminidase activity through steric hindrance and, importantly, effector functions mediated by interactions of the antibody Fc domain with Fc receptors (FcRs) present on effector cells (for example, antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP))18,19,20.

a, Adult humans possess pre-existing immunity to the H1 HA including antibodies and memory B cells with specificity in the stalk domain. Sequential vaccination with cHA constructs that feature head domains from avian influenza virus subtypes but share the same H1 stalk domain may redirect the immune response to the immunosubdominant stalk through selective recall of memory B cell responses. b, Phylogenetic tree of influenza virus HAs. The tree was constructed using amino acid sequences in ClustalOmega and visualized using FigTree. Groups and clades are annotated. H1 (green shade) was used as stalk domain in vaccine constructs; H5 and H8 (shaded in purple) were used as donors for the head domain of the vaccine construct. Breadth of antibody responses was measured using full-length H2, H9 and H18 HAs (blue shade) as well as the H1 stalk. The scale bar represents a 5% difference in amino acid identity. c, Study design including vaccination and sampling time points. Adj., adjuvant; i.m., intramuscular; i.n., intranasal.
The immunodominance of the HA head makes it difficult to induce a potent antibody response against the stalk domain. To redirect the immune response from the head to the stalk domain we have developed a sequential chimeric HA (cHA) vaccination strategy. cHAs consist of group 1 or group 2 stalk domains in combination with head domains from avian influenza virus subtypes21. The two cHA constructs used in this study carried head domains from H8 and H5 HA subtypes combined with an H1 stalk (Fig. 1). Sequential vaccination of animals with cHAs that share the same stalk but have divergent heads induces high antibody titers against the stalk domain and provides broad protection against challenge with divergent influenza viruses22,23,24,25,26. A single vaccination with a cHA in naïve animals induces a primary immune response to the head domain and low-level immune priming against the stalk. Boosting with a cHA that has the same stalk but a different head induces another primary response against the new head but a recall response against the stalk, since the immune system has already seen that stalk. In naïve animals, a third vaccination with a cHA that has yet another head domain but again the same stalk further boosts anti-stalk antibodies22,23,24,25,26. However, most adults are already primed for the stalk through previous exposures, e.g., by natural infection27,28.
The purpose of this study was to evaluate the safety and immunogenicity of group 1 cHA-based universal influenza virus vaccine candidates. The interim results of this trial were previously reported29. Here we report the full study analysis including long-term antibody kinetics up to 18 months after vaccination. This manuscript substantially advances our understanding of the immune responses beyond previously published interim data in terms of longevity of immune responses more than a year after vaccination and functionality including antibody neutralization, ADCC, ADCP, HI and the protection observed in the mouse model after adoptive transfer of sera of vaccinees.
Results
Clinical trial design and vaccine characteristics
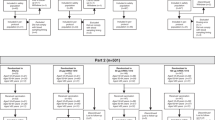
We performed a randomized, multicenter, observer-blind, placebo-controlled phase I clinical trial to determine safety and immunogenicity of cHA-based vaccines (ClinicalTrials.gov identifier NCT03300050). The interim findings of this study (including blinded aggregate vaccine safety) have been previously reported29. Of the 131 subjects screened, 66 were enrolled (1 ineligible subject was randomized in error but replaced before receiving study treatment, resulting in 65 randomized participants) into three different vaccine groups and two placebo control groups (Figs. 1 and 2).

Subject disposition for all volunteers assessed for eligibility is shown. The death in group 3 was caused by a motor vehicle accident and was unrelated to the study vaccine. CCMHC, Cincinnati Children’s Hospital Medical Center; DCRI, Duke Clinical Research Institute.
Group 1 (LAIV8-IIV5/AS03) received an intranasal (i.n.) live-attenuated influenza virus vaccine (LAIV; 107.5 50% egg infectious doses) expressing a cH8/1 HA and an N1 neuraminidase (NA) (H1 stalk and N1 from pandemic H1N1 strain A/California/04/09 for all vaccines) with a backbone from the Leningrad master donor strain as prime on day 1. Individuals in this group were then boosted intramuscularly with an AS03-adjuvanted inactivated influenza virus vaccine (IIV) expressing a cH5/1 HA and an N1 NA on day 85. The rationale for testing a live-attenuated prime strategy was that previous studies in humans using a live-attenuated H5N1 vaccine as a prime had shown superior antibody titers after boost as compared with giving one or two doses of IIV30,31. Also, LAIV often induces mucosal immunity, which would be an advantage. Group 2 (LAIV8-IIV5) was similar but received the IIV boost without adjuvant. Group 3 served as a placebo control for the LAIV-IIV groups and received i.n. saline on day 1 and intramuscular (i.m.) PBS on day 85. Group 4 (IIV8/AS03-IIV5/AS03) received i.m. AS03-adjuvanted cH8/1N1 IIV on day 1 and i.m. AS03-adjuvanted cH5/1N1 at day 85. Group 5 received i.m. PBS on days 1 and 85 and served as placebo control group for the IIV8/AS03-IIV5/AS03 group. For the majority of analyses, groups 3 and 5 were combined (designated Placebo).
Safety
Blinded, aggregate interim safety data for this study have been previously published29. The study has since been completed and unblinded, enabling vaccination-group-specific safety data analyses. All solicited reactions were mild to moderate unless noted. Following a prime dose of i.n. cH8/1N1 LAIV (groups 1 and 2), solicited local reactions (rhinorrhea or nasal congestion) were reported by 12 of 33 (36%) subjects. Rhinorrhea was of 1–4-d duration while nasal congestion lasted 1–6 d. Solicited systemic reactions were reported by 21 of 33 (64%) of the same subjects. The most common reactions were headache in 11 of 33 (33%), fatigue in 7 of 33 (21%), myalgia in 6 of 33 (18%) and nausea in 6 of 33 (18%) subjects. Almost all reactions were of 1–4-d duration, except in one subject who reported multiple severe reactions beginning on day 6, which notably overlapped with adverse events (AEs) of severe cervicitis and gastroenteritis, and which were assessed as being unrelated to the study product. The three control subjects who received i.n. saline (group 3) did not report any solicited local reactions; one of three (33%) reported headache.
Following a prime dose of i.m. cH8/1N1 IIV + AS03 (group 4), solicited local reactions (pain, swelling or induration at injection site) were reported by 11 of 15 (73%) subjects with one reported as severe. One subject also reported concomitant swelling and erythema. Solicited systemic reactions were reported by 10 of 15 (67%), the most common being fatigue, headache and myalgia, each reported by 6 of 15 (40%) subjects. For the ten control subjects who received i.m. PBS (group 5), one (10%) subject reported injection site pain, while four of ten (40%) reported solicited systemic reactions, the most common being fatigue in three of ten (30%) subjects.
Following a booster dose of i.m. cH5/1N1 IIV + AS03 (groups 1 and 4), the only solicited local reaction reported was injection site pain in 18 of 31 (58%) subjects: three of these were reported as severe. Solicited systemic reactions were reported by 15 of 31 (48%), most commonly fatigue in 10 of 31 (32%) and myalgia in 6 of 31 (19%) including three with reported severe myalgia.
Following a booster dose of i.m. cH5/1N1 IIV with no adjuvant (group 2), the only solicited local reaction reported was mild injection site pain in 1 of 13 (8%) subjects. Solicited systemic reactions were reported by 5 of 13 (39%), with the most common reactions being headache in 4 of 13 (31%) and fatigue in 3 of 13 (23%) subjects. One subject reported multiple severe reactions, with a reported concurrent medically attended severe unsolicited AE of viral upper respiratory tract infection.
None of the 12 booster dose control subjects (one participant died in a non-study-related motor vehicle accident before receiving the booster) who received i.m. PBS (groups 3 and 5) reported solicited local reactions. Solicited systemic reactions of headache and fatigue were reported by 1 of 12 (8%) subjects.
Unsolicited AEs were reported in 12 of 19 (63%), 10 of 14 (71%) and 8 of 15 (53%) subjects in groups 1, 2 and 4, respectively, and in 2 of 3 (67%) and 6 of 10 (60%) subjects in control groups 3 and 5, respectively. Unsolicited AEs of grade 2 or greater and assessed as related to study product were reported in 1 of 19 (5%) subjects in group 1 (night sweats), 2 of 14 (14%) subjects in group 2 (nasal mucosal disorder and hyperhidrosis) and 1 of 15 (7%) subjects in group 4 (lymphadenopathy), and all were reported as moderate in severity.
During the study, three unrelated serious adverse events (SAEs) were reported, all in placebo control recipients, including a motor vehicle accident, a spontaneous abortion and an upper gastrointestinal bleeding. Figure 2 specifies the inclusion of individuals into immunology and safety analyses. No notable safety signals regarding clinical safety laboratory test results were detected, and there were no reported potential immune-mediated diseases (pIMDs).
LAIV shedding and influenza-like illness (ILI) follow-up
Of the 33 subjects in groups 1 and 2 tested for influenza A virus shedding following the prime dose of LAIV by reverse transcription PCR (RT–PCR), positive tests were observed for 11 subjects on day 2, two subjects on day 3, no subjects on day 4 and three subjects on day 5. The reference laboratory confirmed vaccine virus for 5 of 11 of the day 2 positives, 2 of 2 of the day 3 positives and 0 of 3 of the day 5 positives using a cH8/1N1 specific RT-PCR (Supplementary Table 1). None of the PCR-positive specimens were positive by virus culture. No viral shedding was detected in group 3 subjects. During two consecutive winter seasons following vaccination, there were 17 episodes of ILIs in 12 subjects in all groups except for group 4. Of the 17 episodes, two were positive for influenza A (in group 1 and group 2) and one for influenza B (group 3). There was no evidence for an increase in frequency or severity of ILI in the study subjects. No influenza cases were identified in groups 4 and 5 (Supplementary Table 2).
Inactivated, adjuvanted cHA vaccines induce strong anti-HA stalk responses in serum
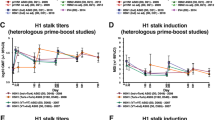
The primary immunological readout for this study was the level of anti-HA stalk antibodies as measured by enzyme-linked immunosorbent assays (ELISAs) with a chimeric 6/1 HA (cH6/1) protein as the substrate. This substrate should be recognized by anti-H1 stalk antibodies, but since humans are naïve to the H6 head domain, few anti-head antibodies will be detected. Initial ELISAs were performed by a contract research organization (NEOMED-LABS). Grouped aggregate data up to day 113 after prime have been previously discussed for this assay29. In this manuscript, we have now included individual data and additional time points up to day 420 to assess antibody persistence. The assay developed at NEOMED-LABS uses ELISA units (EUs) as a readout and is a qualified assay (that is, the assay has been developed and tested according to good laboratory practice (GLP) methods, but has not yet undergone formal GLP assay validation studies) that utilizes high coating concentrations of antigen and therefore likely also detects low-affinity antibodies. As previously described, a single vaccination with LAIV did not increase serum IgG titers against the stalk. (Fig. 3a). However, when LAIV-primed individuals were boosted with IIV5/AS03, they induced a robust anti-stalk response. When the booster dose was given without the adjuvant (IIV5), a lower induction of anti-HA stalk antibodies was observed and the response was more heterogeneous between the vaccinees. Priming with IIV8/AS03 induced a very strong antibody response against the stalk (Fig. 3a). Serum antibody titers waned slightly between day 29 and day 85 but then increased again after IIV5/AS03 vaccination to levels similar to day 29. Importantly, the new long-term follow-up data show a slight decline between day 113 (28 d after boost) and day 252, but no further decrease in the IIV8/AS03-IIV5/AS03 group between day 252 and day 420 (titers persisting at approximately 2.25-fold above baseline) (Fig. 3b). While titers also appeared to stabilize in the LAIV8-IIV5/AS03 and LAIV8-IIV5 groups, they did so at a lower level (Fig. 3b). There was no increase of anti-stalk antibody titers measured for the placebo groups in this assay over time (Fig. 3a,b).

a, Serum anti-stalk IgG titers in the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups as measured against recombinant cH6/1 HA substrate in a qualified assay with high substrate coating concentration at NEOMED-LABS. Faint lines indicate reactivity of different individuals; bold lines indicate GMTs of the respective groups. Biologically independent samples; group 1, n = 19; group 2, n = 14; group 4, n = 15; groups 3 + 5, n = 13 (3 + 10); examined in one independent experiment. b, Geometric mean fold induction of antibody titers based on data in a. c,d, Data in c (antibody titers) and d (induction) show the same (including n) but as performed with an ELISA that uses a low coating concentration and favors high-affinity antibodies. e,f, Data from the same groups (same n as listed above) for serum IgA reactivity against the H1 stalk with titers shown in e and induction shown in f. Data shown in c and f include an additional time point (day 588).
To study the HA stalk-specific antibody responses in more detail, ELISAs were also performed using a previously established protocol, using the same substrate and including an additional time point (day 588). This assay protocol was previously used in a human cohort study that showed correlation with protection17 and therefore makes a more direct comparison possible. The assay uses a lower coating concentration, which should result in less binding of low-affinity antibodies. In general, very similar antibody induction patterns were observed for all groups in this assay (Fig. 3c). The baseline values were lower, likely due to the reduced detection of low-affinity antibodies, resulting in a higher-fold induction over baseline (Fig. 3d). Since antibody longevity is highly important for a supra-seasonal influenza virus vaccine, the long-term persistence data up to 1.5 yr after vaccination measured in this assay were encouraging (approximately 4.3-fold above baseline at day 588 in the IIV8/AS03-IIV5/AS03 group) (Fig. 3c,d).
Since IgA antibodies are important for protection against respiratory viruses, additional testing was performed to measure HA stalk-specific IgA antibodies. The IgA levels in serum were lower at baseline than the IgG levels, which is consistent with previous findings27 (Fig. 3e). After vaccination, the induction of IgA was also lower compared with IgG, with a maximum fold induction of 4.7 after the prime in the IIV8/AS03-IIV5/AS03 group. However, IgA levels remained similar to peak titers up to day 420, especially in the IIV8/AS03-IIV5/AS03 group (Fig. 3e,f).
There was no indication that LAIV priming had a positive effect on serum IgA titers. However, we also assessed antibody induction at mucosal surfaces since LAIV is known to induce mucosal antibodies32. Saliva samples were analyzed in ELISAs for anti-stalk IgA, secretory IgA (sIgA) and IgG using specific secondary antibodies. No induction of IgA or sIgA was detected (Supplementary Fig. 1a–d). A low signal for anti-stalk IgG was detected after IIV8/AS03 priming and IIV5/AS03 boost, likely due to transudation of IgG from serum (Supplementary Fig. 1e,f).
Since antibodies that can bind head domains of different HA subtypes have been reported at low levels in humans33,34, we also tested the serum samples in an ELISA against a trimeric ‘Mini HA’ construct containing parts of the HA stalk domain that feature cross-reactive epitopes, but that lacks a head domain. Binding profiles detected with the Mini HA construct were similar to the ones measured in the cH6/1 stalk ELISAs and the results from the two assays showed strong correlation (Supplementary Fig. 2). Slightly lower antibody levels were detected using the Mini HA probe compared with the cHA antigen, which could be due to antibodies binding conserved epitopes in the HA head or head–stalk interface. Furthermore, we investigated whether the antibodies elicited after vaccination compete with a known broadly cross-reactive HA stalk-specific monoclonal antibody (CR9114)16. The five highest HA stalk antibody-inducing individuals (as measured by cH6/1 ELISA) were selected from each group to compare competition at baseline with day 113 after vaccination. We observed an increase in competition against the HA stalk-specific antibody after vaccination for all groups, except for placebo recipients, indicating that antibodies binding similar epitopes to CR9114 were induced by cHA vaccination (Supplementary Fig. 3).
Lastly, we also tested the effect on antibody affinity. While measuring apparent affinities is not possible in the context of polyclonal serum, the dissociation constant (Koff) can be used as a substitute for polyclonal avidity35. We performed measurements on a biolayer interferometer with cH6/1 HA on the sensor and found that the Koff decreased (which means avidity increased) after adjuvanted IIV vaccinations while LAIV8 and nonadjuvanted IIV5 had no effect (Supplementary Fig. 4). However, it cannot be excluded that total HA stalk-specific antibody levels are responsible for the observed decrease in Koff rates.
The induced antibody response is broad within group 1 HAs
To measure the breadth of the elicited immune responses, antibody titers against H2, H9 and H18 were tested (all group 1; Fig. 1b). Additionally, ELISAs were performed against H3 HA (group 2). The grouped aggregate data up to day 113 were previously published for H2, H9 and H18 (but not for H3)29. As previously described, antibody responses against the heterologous group 1 HAs roughly followed the patterns observed for HA stalk-specific antibodies, with lower responses for the antigenically more distantly related HAs, H9 and H18 (Fig. 4a–f). Importantly, the additional longevity data show that the broad responses are also maintained at least up to day 420 after vaccination.

a,c,e,g, Serum antibody titers against H2 (a), H9 (c), H18 (e) and H3 (g) in the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups as measured against the respective recombinant HA substrates. Faint lines indicate reactivity of different individuals; bold lines indicate GMTs of the respective groups. Biologically independent samples; group 1, n = 19; group 2, n = 14; group 4, n = 15; groups 3 + 5, n = 13 (3 + 10); examined in one independent experiment. Geometric mean fold induction of antibody titers based on data in a, c, e and g is shown in b, d, f and h, respectively.
As expected, no increases in reactivity to H3 HA (group 2) were found in any of the groups except for the IIV8/AS03-IIV5/AS03 group, where a small increase was seen after the prime (Fig. 4g,h). The H3 results align well with results from animal models, human monoclonal antibody reactivity profiles and serological studies36,37,38,39.
cHA vaccination induces multifunctional antibodies
We next aimed to dissect the functionality of cHA vaccine-elicited antibodies. Fc-mediated functions have been previously shown to be important for protection conferred by HA stalk-specific antibodies18,40. We tested these Fc-mediated functionalities in ADCC and ADCP reporter assays. The patterns of stalk-mediated ADCC-active antibodies measured by the reporter assay followed those measured in the cH6/1 ELISAs (Fig. 5a,b), and the ADCP assay data also followed the same trend (Supplementary Fig. 5).

a, Serum anti-stalk ADCC activity in the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups as measured in a reporter assay. Biologically independent samples; group 1, n = 19; group 2, n = 14; group 4, n = 15; groups 3 + 5, n = 13 (3 + 10); examined in one independent experiment. b, Geometric mean fold induction of ADCC reporter activity based on data from a. c, NA-based serum NI activity in the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups as measured in a neuraminidase inhibition assay. Biologically independent samples; group 1, n = 19; group 2, n = 14; group 4, n = 15; groups 3 + 5, n = 13 (3 + 10); examined in one independent experiment. d, Geometric mean fold induction of NI activity based on data from a. e, HA stalk-based serum NI activity through steric hindrance in the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups as measured in a neuraminidase inhibition assay. Biologically independent samples; group 1, n = 19; group 2, n = 14; group 4, n = 15; groups 3 + 5, n = 13 (3 + 10); examined in one independent experiment. f, Geometric mean fold induction of NI activity based on data from e. Faint lines indicate reactivity of different individuals; bold lines indicate GMTs of the respective groups. IC50, half-maximum inhibitory concentration.
We next tested whether neutralizing activity would increase after cHA vaccination. First, to primarily measure HA stalk-reactive antibodies, microneutralization (MN) assays were performed with a cH6/1N5 virus. This virus expresses a mismatched HA head and NA to which humans are naïve, but a conserved HA stalk domain. Neutralization titers increased approximately twofold after prime or boost with IIV vaccine in the vaccination groups while no increase was seen in the placebo groups (Extended Data Fig. 1). We further tested neutralization of a panel of heterologous virus strains. The neutralization results obtained for these viruses were similar, with lower induction of neutralization activity observed with a pandemic H1N1 virus (Extended Data Fig. 1).
HI assays were performed with the same virus panel. Interestingly, some induction of HI-active antibodies was detected, potentially targeting conserved epitopes in the HA head domain (Extended Data Fig. 2). The HI assay was also performed for the cH8/1N1 and cH5/1N1 vaccine strains. Of note, the initial cH8/1N1 vaccination seemed to elicit head-specific responses that could also be detected in HI assays using the cH5/1N1 vaccine strain (Extended Data Fig. 2). The cH5/1N1 vaccination boosted homologous HI responses, but did not reciprocally further increase cH8/1N1 HI activity. To investigate antibody responses against conserved epitopes in the HA head domain, we performed a competition ELISA with a previously published antibody that binds to a conserved epitope in the HA trimer interface (D1 H1-3/H3-3)33. The assay was performed using the same subset of participants as described above for the CR9114 competition ELISAs. Particularly in the groups that received AS03-adjuvanted vaccinations, we saw an increase in competition against antibodies recognizing the HA trimer interface (Supplementary Fig. 6). These data indicate that cHA vaccination also elicits antibodies against conserved epitopes in the HA head domain, which may contribute to protection from infection41,42.
Further, to assess antibodies that inhibit NA, we performed exploratory neuraminidase inhibition (NI) assays with an H7N1 reassortant virus. No antibodies against group 2 HAs (for example, H7) that could interfere with NA activity were induced by the vaccine; therefore, the titers measured with the H7N1 virus likely reflect true binding to NA. Titers followed a similar pattern as seen for the anti-stalk ELISA titers. The titers increased after IIV vaccinations, more so after adjuvanted IIV than after nonadjuvanted IIV vaccination, with the strongest peak induction in the IIV8/AS03-IIV5/AS03 group after the prime (Fig. 5c,d). The titers then declined slowly to 2.1-fold above baseline in the IIV8/AS03-IIV5/AS03 group. In the other groups, the titers returned to baseline values. ELISA titers to NA followed similar trends (Supplementary Fig. 7). NI can also be caused by steric hindrance of the interaction between NA and substrate by HA stalk-binding antibodies20. To assess the titers induced by this mechanism, we used an H6N3 virus. Anti-stalk antibodies bind to the H6 HA, which is a group 1 HA (Fig. 1b), but N1 vaccination generally does not induce titers to N3 (and humans are naïve to N3 NA). Again, the pattern followed the titers measured in the anti-stalk ELISAs, implying that HA stalk-based NI-active antibodies are induced (Fig. 5e,f).
Anti-stalk antibodies induced by vaccination confer protection in a serum transfer study
While serological assays are well suited to characterize different aspects of the immune response to vaccination, they do not necessarily indicate that in vivo protection was achieved. We therefore turned to a well-established in vivo model in which serum is transferred into mice that are then challenged with a virus of choice40,43. We transferred pools of prevaccination, postboost (day 113) or day 420 serum from each trial group into sets of mice and challenged them with a cH6/1N5 virus which, as described above, should only be recognized by stalk-reactive antibodies in human serum. The challenge dose was determined by performing the same experiment with commercially available human serum. The sera used for the dose-finding experiment were collected from de-identified healthy donors in 2019 and were presumed to have similar pre-existing antibody titers as the vaccination groups (unblinded ELISA data were not available when the experiment was performed). A challenge dose that induced severe weight loss in the presence of serum (200 plaque-forming units (p.f.u.)) was then chosen for subsequent passive transfer/challenge experiments (Supplementary Fig. 8). For the LAIV8-IIV5/AS03 group, mice that received day 1 serum pools lost substantial amounts of weight after challenge, while mice that received pooled postboost serum from this group (day 113) showed no weight loss (Fig. 6a). Mice receiving day 420 serum showed less morbidity than mice that received prevaccination serum but more when compared with mice that received the day 113 serum pool. Patterns for the LAIV8-IIV5 group were similar but the protective effect of the day 113 serum was lower, as expected based on serology. Animals that received IIV8/AS03-IIV5/AS03 samples were protected by day 113 and day 420 serum. However, the baseline protection of the IIV8/AS03-IIV5/AS03 serum pool was also higher, likely due to the contribution of serum from some individuals who had already high stalk titers on day 1 within this group (Fig. 3). Animals receiving serum pools from the placebo groups showed weight loss at all time points, although the group that received day 420 serum seemed to recover faster. By calculating the decrease in weight loss observed in mice that received postvaccination sera compared with baseline sera, improved protection in cHA vaccination groups, but not in the placebo group, can be observed (Fig. 6b). An additional experiment was performed (using a lower 10 p.f.u. for infections, since differences in lung titers can often not be measured when using high challenge doses) assessing viral lung titers on days 3 and 6 after challenge instead of weight loss. There was substantial variation in measured viral lung titers within all groups, but an overall trend towards lower viral titers in mice receiving day 113 serum from cHA-vaccinated individuals was observed (Supplementary Fig. 9). The reduction in lung titers was less substantial than the observed protection from weight loss, which has been previously observed for this specific challenge virus43. This seems to indicate that complete neutralization of this virus may not easily be accomplished by HA stalk antibodies in this in vivo model, but they might still protect through modulating the cytokine environment in the lung44.

a, Weight loss of mice (n = 10 per group; except placebo group day 1 and day 113, n = 9) that received day 1, day 113 (postboost) or day 420 serum pools from the LAIV8-IIV5/AS03, LAIV8-IIV5, IIV8/AS03-IIV5/AS03 and the combined placebo groups and were then challenged with a cH6/1N5 virus to assess stalk antibody-based protection in one independent experiment. Shaded areas represent s.e.m. b, The difference in weight loss of mice (n = 10 per group, except placebo group day 1 and day 113 n = 9) receiving postvaccination serum versus baseline serum in each group was calculated, respectively. Bars show the mean and error bars show s.e.m.
In a previous family cohort study, no human subjects with stalk antibody area under the curve (AUC) levels exceeding 1,280 were found to be infected by H1N1 virus17. Based on this presumably protective level, we assessed the percentage of individuals that would potentially be protected in each group per time point. It should, however, be noted that the HA stalk antibodies of individuals in the earlier study were elicited by natural infection and contribution of other immune mechanisms to protection cannot be completely excluded. The percentage of individuals with titers above 1,280 AUC was low in all groups at day 1 (6.7–14.3%), as expected (Supplementary Fig. 10). For the LAIV-IIV groups, the percentages increased to 93.8% and 30.8% after the IIV5/AS03 and IIV5 booster doses, respectively, and then declined to levels between 7.7% and 40.9% between days 242 and 588. Levels for IIV8/AS03-IIV5/AS03 increased to 93.5% after the prime and to 100% after boost. They stayed constant at levels of 66.7–73.3% between days 242 and 588, indicating potential long-term protection.
Discussion
In this study, we tested our vaccine candidates in healthy adults, who were primed for group 1 stalk antibodies due to previous exposures via natural influenza virus infections and vaccinations as evidenced by the measured baseline titers27. We found that vaccination with adjuvanted, inactivated cHA vaccines induced remarkably high anti-stalk antibody titers even after a single administration. The titers achieved after one vaccination exceeded anti-stalk titer levels at which protection from pandemic H1N1 virus infection was observed in a Nicaraguan family cohort study17. IgG serum antibody titers were long lived, especially in the IIV8/AS03-IIV5/AS03 group. While the titers decreased after vaccination, they stabilized well over baseline at 6 months after boost and remained constant even 18 months after boost. However, it remains to be seen if HA stalk antibodies elicited by vaccination will be equally protective compared with antibodies elicited by natural infection.
The induced antibody responses also showed functional activities. Fc-FcR-mediated effector functions are an important mechanism of protection of anti-stalk antibodies in vivo in animal models18. Antibodies induced in our trial showed strong activity in both ADCC and ADCP reporter bioassays, which correlate with functional ADCC assays45. Neutralizing and even HI activities were also induced by the cHA vaccines, albeit at a low level. However, these functions are a major contributor to traditional protection mediated by seasonal influenza virus vaccines and might also play a role in the protection provided by novel vaccination approaches. In addition, we could show both HA-stalk-based20 and NA-based NI. Finally, passive transfer of postvaccination serum protected mice from challenge as compared with prevaccination serum. These antiviral activities reflect the multi-functionality of the characterized anti-stalk monoclonal antibodies19,20.
The tested LAIV prime did not induce any measurable antibody responses in serum or on mucosal surfaces and did not have a positive impact on antibody durability. It is possible that a higher dose of LAIV could have resulted in higher mucosal antibody responses. It is very likely that the LAIV8 vaccine strain did not replicate in the nasal mucosa and therefore did not contribute to the immune response. Potential reasons for this could be the avian receptor specificity of the H8 head, which might have dampened virus replication in the upper respiratory tract, as well as potential hyper-attenuation of cHA-expressing LAIVs in humans.
Another interesting observation was that the titers after the boost with IIV5/AS03 did not exceed the titers after the initial IIV8/AS03 vaccination in the IIV8/AS03-IIV5/AS03 group. Potential explanations could be that the titers had reached a ceiling after the first vaccination, that a nonoptimal interval was chosen between prime and boost, antigen masking by existing antibodies or that the two vaccines (IIV8 versus IIV5) had a different intrinsic immunogenicity. While not always exceeding the initial postprime peak antibody responses, an increase from preboost antibody titers to postboost antibody titers was detected in almost all assays and the second vaccination seemed to have a positive impact on the longevity of the antibody response.
Even though reactogenicity findings were relatively common and somewhat confounded by concurrent unrelated AEs, all of the study vaccines were found to have acceptable solicited AE profiles comparable to similar approved vaccines46. As expected, AS03-adjuvanted vaccines were associated with pain at the injection site, while LAIV vaccines induced mild nasal symptoms.
In summary, we show that vaccination with inactivated adjuvanted split virion cHA vaccines induces strong, broad, long-lasting and functional anti-stalk antibodies and appears to have acceptable safety in adults. While this clinical trial represents a proof of principle in humans, it only focused on group 1 HAs. Development of group 2 cHA22,47 and influenza B mosaic HA48 vaccine candidates is currently ongoing. Combining these constructs into a trivalent vaccine may enable protection against all drifted seasonal, zoonotic and emerging pandemic influenza viruses. While much more work is needed to further develop this vaccine concept, this first-in-human study supports further development of truly universal influenza virus vaccines.
Methods
Vaccines and vaccination
Two different vaccine platforms were utilized for this trial (ClinicalTrials.gov identifier NCT03300050). First, an LAIV carrying the cH8/1 HA (head domain from A/mallard/Sweden/24/02 (H8N4), stalk domain from A/California/04/09 (Cal09, H1N1)), an N1 NA from Cal09 and the internal genes of the A/Leningrad/134/17/57 (refs. 49,50,51) was rescued using reverse genetics as described24. This vaccine was manufactured in embryonated chicken eggs at Meridian Life Sciences in Memphis, Tennessee, and formulated in sterile saline. Two IIV vaccines, one carrying the same cH8/1 HA and one carrying a cH5/1 HA (head domain from A/Vietnam/1203/04 (H5N1), stalk domain from Cal09), were also rescued with the same N1 NA in the A/Puerto Rico/8/34 (PR8, H1N1) backbone and manufactured in embryonated chicken eggs by GSK as described elsewhere24,29.
LAIV was administered intranasally at 107.5 50% egg infectious doses. Vaccinees had to blow their noses and lay supine with their heads tilted backward, followed by the dropwise administration of 0.25 ml per nostril via a needle-less syringe. The vaccinees were also asked not to sneeze during the procedure to avoid expelling parts of the vaccine solution from their nose. Sterile saline was blindly administered in the same way to a control group. Administration of the LAIV or sterile saline was carried out in a containment unit and vaccinees were required to stay in the containment unit for at least 5 d after vaccination or until they were confirmed to be virus-negative by quantitative RT-PCR of nasal and oropharyngeal swabs on 3 consecutive days (cut-off was a cycle threshold value of ≤40). Results from the RT-PCR were only accessible to a designated unmasked person to keep the study observer-blind and results were only communicated if they would delay release from the containment unit. Any RT-PCR-positive swabs were also cultured on Madin-Darby canine kidney (MDCK) cells followed by immunostaining of the cells with an H8 head-specific monoclonal antibody (monoclonal antibody 1A7 (ref. 52)). IIV was administered intramuscularly in a volume of 0.5 ml with either PBS or AS03A. The antigen content was 15 μg of the respective HA (cH5/1 or cH8/1). PBS was administered to control groups.
Three different vaccination regimens as well as two placebo groups were included in the trial. Group 1 received cH8/1N1 LAIV on day 1 followed by AS03A-adjuvanted cH5/1N1 IIV on day 85 (LAIV8-IIV5/AS03). Group 2 received the same vaccination regimen but with the booster vaccination nonadjuvanted (LAIV8-IIV5). Group 3 was the inpatient control group and received normal saline intranasally followed by PBS intramuscularly (SALINE-PBS). Group 4 received adjuvanted cH8/1N1 IIV followed by adjuvanted cH5/1N1 IIV (IIV8/AS03-IIV5/AS03). Group 5 served as outpatient placebo group and received PBS intramuscularly twice (PBS-PBS). For many of the immunological assays, the SALINE-PBS and PBS-PBS groups were pooled and designated as placebo.
Study design
Methods for this randomized, placebo-controlled, observer-blind, phase I clinical study conducted at Cincinnati Children’s Hospital Medical Center (CCHMC, Cincinnati, OH, USA) and Duke Early Phase Clinical Research Unit (Durham, NC, USA) have been previously described by Bernstein et al.29. Briefly, 66 participants were block-randomized (stratified by site) in a ratio of 4:3:1:3:2 to receive LAIV8-IIV5/AS03, LAIV8-IIV5, SALINE-PBS, IIV8/AS03-IIV5/AS03 or PBS-PBS. One ineligible subject was randomized in error but replaced before receiving study treatment, resulting in 65 randomized participants. The CCHMC Institutional Review Board (IRB) served as the central IRB of record for review, approval and oversight of this study on behalf of the Icahn School of Medicine at Mount Sinai (ISMMS) IRB, Duke IRB and PATH Research Ethics Committee. Written, informed consent was obtained from all study participants. Drs. Ruth Karron, Kathleen Neuzil, Hanna Nohynek and Stephen Thomas served as the Independent Data Monitoring Committee for this clinical trial. ClinicalTrials.gov identifier NCT03300050.
Changes to design/outcomes
The study was originally planned to end at day 420 (12 months after booster dose) but was extended an additional 6 months for the collection of specimens at day 588 (18 months after booster dose) to measure the persistence of the immune response. All analyses except one noted below are based on the statistical analysis plan, which was amended to accommodate additional endpoints at day 588. Additional exploratory assays including low-protein-coating ELISAs, NI assays, Koff measurements and Mini HA ELISAs were not defined in the study protocol.
Participants
Eligible participants included men and women aged 18–39 yr. To be included, participants needed to be healthy and of nonchildbearing potential, sterile or willing to practice adequate contraception from the first vaccination to 2 months after the booster dose. Additional eligibility criteria were described by Bernstein et al.29 and a full list of inclusion and exclusion criteria is provided at https://clinicaltrials.gov/ct2/show/NCT03300050. The first enrollment occurred on 10 October 2017 and the last subject completed the trial on 9 August 2019.
Outcomes
The primary objective of this study was to assess safety of the regimens through 28 d after booster (day 113). Secondary and tertiary objectives assessed safety and immunogenicity through 18 months after booster. Primary and secondary objectives through 28 d after booster for safety, ELISAs measuring anti-H1 stalk, anti-H2, anti-H9 and anti-H18 IgG antibodies, and plasmablast and B cell response have been previously described by Bernstein et al.29.
Secondary safety objectives after day 113 reported here include hematological and biochemical laboratory abnormalities up to 12 months after boost and frequency of medically attended events, laboratory-confirmed ILIs, pIMDs and SAEs up to 18 months after boost in each study group.
Secondary immunogenicity objectives not reported previously include seropositivity rates, geometric mean titers (GMTs), percentages of subjects with a fourfold or greater increase in titer, percentages of subjects with a tenfold or greater increase in titer and mean geometric increases of IgG targeting H1 stalk, H2, H9 and H18 as measured by ELISAs from 28 d after booster (day 113) to 12 months after booster (day 420), ELISAs measuring anti-N1 IgG, anti-H3 IgG and anti-H1 stalk IgA antibodies from day 1 through 12 months after booster (day 420), and microneutralization assays using cH6/1N5 virus, pH1N1 virus, asH1N1 virus and H5N8 virus from day 1 through 12 months after booster. ADCC activity was also evaluated from day 1 through 12 months after booster.
Tertiary immunogenicity objectives include evaluation of cell-mediated immune responses after each vaccination, evaluation of biomarkers associated with different vaccination regimens by transcriptomic analysis, evaluation of the immune response by HI and evaluation of ADCP.
Interim analyses and stopping guidelines
As described by Bernstein et al., an interim analysis occurred 28 d after booster (day 113) of blinded safety data and a subset of immunogenicity results29.
Safety evaluation
Following administration of investigational products, subjects were provided with diaries to record any local or systemic solicited AE for 7 d and any unsolicited AE for 28 d. Solicited local reactions included rhinorrhea and nasal congestion for investigational product administered intranasally and pain or tenderness, induration or swelling, and erythema at the site of injection for investigational product administered intramuscularly. Solicited general reactions for all subjects included arthralgia, cough, fatigue, fever, gastrointestinal symptoms (nausea, vomiting, diarrhea or abdominal pain), headache, myalgia, shivering, sore throat and wheeze.
Subjects were also monitored during two influenza transmission seasons for any protocol-defined ILI and throughout the duration of the trial for any pIMD, AE leading to withdrawal from the study, medically attended AE or SAE. At each study visit, venous blood was collected for a complete blood count and to measure serum creatinine, urea nitrogen, and alanine and aspartate transaminase levels.
Immunological analysis overview
Samples for immunological analysis were taken on day 1 (saliva, serum, whole blood and peripheral blood mononuclear cells (PBMCs)), day 3 (whole blood), day 8 (whole blood and PBMCs), day 29 (saliva, serum, whole blood and PBMCs), day 85 (saliva, serum, whole blood and PBMCs), day 92 (whole blood and PBMCs), day 113 (saliva, serum, whole blood and PBMCs), day 252 (saliva, serum, whole blood and PBMCs), day 420 (saliva, serum and PBMCs) and day 588 (serum). The assays performed on different samples are described in detail below.
Cells, viruses and proteins for immunological assays
MDCK cells were grown in DMEM (Gibco) containing penicillin/streptomycin antibiotics (100 U ml−1 penicillin, 100 μg ml−1 streptomycin; Gibco) and fetal bovine serum (FBS, 10%; Corning), resulting in complete DMEM (cDMEM). Human embryonic kidney (293T) cells were grown and maintained in cDMEM. BTI-TN-5B1-4 (High Five, Trichoplusia ni) cells were grown in serum-free Express Five medium (Gibco) supplemented with an antibiotic mix (100 U ml−1 penicillin, 100 μg ml−1 streptomycin) and10% l-glutamine (200 mM; Gibco). Sf9 (Spodoptera frugiperda) cells were maintained in Trichoplusia ni Medium-Formulation Hink (TNM-FH; Gemini Bio-Products) in the presence of 10% FBS and penicillin/streptomycin antibiotics mix.
Influenza A viruses were grown in 8–10-d-old embryonated chicken eggs (Charles River Laboratories) at 37 °C for 2 d. Virus reassortants were rescued by plasmid-based reverse genetic techniques as previously described53. A reassortant virus harboring the H6 head domain from A/mallard/Sweden/81/02 (H6N1), the H1 stalk domain from A/California/04/09 (pandemic H1N1) and N5 from A/mallard/Sweden/86/03 (H12N5) was rescued in combination with the six internal segments from A/Puerto Rico/8/34 (PR8, H1N1). The HA and NA of A/H5N8 (A/gyrfalcon/Washington/41088-6/14), an avian–swine lineage H1N1 influenza virus (A/swine/Jiangsu/40/11, asH1N1) and an H6N3 (HA from A/mallard/Sweden/81/02, NA from A/swine/Missouri/4296424/06) were rescued in a PR8 backbone, respectively. For reassortant H7N1 virus, the HA of A/H7N9 (A/Hunan/02285/2017) and the NA of A/H1N1 (A/Michigan/45/2015) were combined with the internal segments of PR8 virus. IVR-180, which expresses the glycoproteins of A/Singapore/GP1908/15 (pH1N1) and the internal proteins of a high-yield vaccine strain (A/Texas/1/77 (H3N2)), was sourced from the National Institute for Biological Standards and Control and grown as described above.
Recombinant proteins (cH6/1 (H6 head domain from A/mallard/Sweden/81/2002, H1 stalk domain from Cal09 with a stabilizing mutation54), N1 (Cal09) and H3 (H3 from A/Hong Kong/4801/2014)) were expressed in the baculovirus expression system as previously described in detail55. Recombinant proteins for assays at NEOMED-LABS, including cH6/1 protein and wild-type HAs from A/mallard/Netherlands/5/99 (H2N9), A/chicken/Hong Kong/G9/97 (H9N2) and A/flat-faced bat/Peru/033/10 (H18N11), were also expressed in a similar way. Recombinant, stabilized headless H1 protein, Mini HA #4900 (ref. 56), was produced in Expi293F mammalian suspension cells (Life Technologies) as described previously57.
ELISA
Initial per-protocol serum IgG ELISAs using cH6/1 (with d1, d28, d85, d113, d252, d420 samples), H2 (d1, d28, d85, d113, d420), H9 (d1, d28, d85, d113, d420) and H18 (d1, d28, d85, d113, d420) recombinant HA substrates were performed by NEOMED-LABS (Laval, Canada) using a qualified assay according to standard operation procedures (SOPs). Results were expressed as EUs per ml. Briefly, the antigens were coated on 96-well plates at concentrations of 6 µg ml−1 (600 ng per well). After overnight incubation at 4 °C, plates were washed three times (PBS pH 7.4, 0.05% Tween 20), followed by blocking for 1 h at room temperature using 1% polyvinyl alcohol (PVA) in PBS. Plates were washed three times and samples, controls and standard were twofold serially diluted (starting dilutions 1:50 or 1:2,000) in 1% PVA in PBS. After washing three times, a mouse anti-human IgG horseradish peroxidase (HRP) antibody (clone JDC-10, Southern Biotech) was added at a dilution of 1:2,000 in 1% PVA in PBS. Plates were washed five times, developed for 30 min with 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) substrate and stopped using 2 M H2SO4. Plates were read at 450–520-nm wavelengths and EUs were calculated based on interpolation with the serum standard. Positive and negative controls were developed in addition to an antigen-specific standard. The assay cut-off was 66 EU ml−1 for H1, 22 EU ml−1 for H2 and 43 EU ml−1 for H18 (lowest limits of quantification computed using lower limits of precision and lower limits of standard curve accuracy).
Additional ELISAs were performed at ISMMS with blinded samples. For cH6/1 (d1, d28, d85, d113, d252, d420, d588) and Mini HA (d1, d28, d85, d113, d420) serum IgG ELISAs, microtiter 96-well plates (Thermo Fisher) were coated with 50 µl of recombinant cH6/1 HA or Mini HA construct at a concentration of 2 µg ml−1 in PBS (pH 7.4; Gibco) overnight at 4 °C. The following day, the plates were blocked with 220 µl of blocking buffer (PBS supplemented with 0.1% Tween 20 (PBS-T), 3% goat serum (Life Technologies) and 0.5% milk powder (AmericanBio)) for at least 1 h at 24 °C. Human serum samples were diluted to a starting concentration of 1:100 and serially diluted 1:2 in blocking solution. The plates were incubated for at least 2 h at room temperature and washed three times with PBS-T using an automatic plate washer (Molecular Devices). Next, 50 µl of secondary antibody (anti-human IgG conjugated with HRP produced in goat (Sigma)) diluted 1:3,000 in blocking solution was added to each well and incubated for 1 h at room temperature. The plates were washed four times with PBS-T and developed with SigmaFast o-phenylenediamine dihydrochloride (OPD; Sigma) for 10 min, and the reaction was stopped with 3 M HCl (Thermo Fisher). The plates were read at a wavelength of 490 nm with a microplate reader (BioTek) and the data were analyzed in Microsoft Excel and GraphPad Prism 7. The cut-off value defined as an optical density of 0.075 and the AUC values were determined using the AUC calculation function of GraphPad Prism (https://www.graphpad.com/guides/prism/7/statistics/stat_area_under_the_curve.htm). The reciprocal dilution values were used for AUC calculation.
Additional ELISAs for anti-N1 (d1, d28, d85, d113, d420) and anti-H3 (d1, d28, d85, d113, d420) serum IgG; cH6/1 serum IgA (d1, d28, d85, d113, d420); as well as saliva anti-cH6/1 IgG, IgA and sIgA (d1, d28, d85, d113, d420 in all cases) were run on blinded samples according to SOPs as follows. Briefly, ultra-high-binding polystyrene 96-well plates (Immulon 4HBX, Thermo Scientific) were coated with 100 μl per well of recombinant protein in PBS solution (Gibco) at a concentration of 6 μg ml−1. After a 12-h incubation period at 4 °C, plates were washed with PBS-T using an automated plate washer system (AquaMax 2000, Molecular Devices). Plates were blocked for 1–2 h with 220 μl per well of blocking solution consisting of PBS-T, 3% goat serum (Gibco) and 0.5% nonfat dry milk (AmericanBio). Serum/saliva samples were serially diluted (twofold dilutions in blocking solution) starting from initial dilutions of: 1:100 for cH6/1 IgA in serum; 1:800 for N1 and H3 IgG in serum; and 1:4 for cH6/1-specific IgA and sIgA in saliva. Diluted samples were added to the plates (100 μl per well), followed by incubation at room temperature for 2 h. Plates were washed as described before, and the respective secondary antibodies were diluted in blocking solution and added to the plates (50 μl per well) at the indicated dilutions: Goat Anti-Human IgA α-chain-specific HRP (Sigma), 1:24,000 for serum cH6/1 IgA and 1:750 for saliva cH6/1 IgA; Goat Anti-Human IgG Fc-specific HRP (Sigma), 1:3,000 for saliva cH6/1 IgG; and Goat Anti-Human secretory component HRP (Nordic-MUbio), 1:750 for saliva sIgA. Plates were incubated for 1 h at room temperature and washed as described before. Substrate TMB (Bio-Rad) was added to the plates (100 μl per well) and the reaction was left to proceed for 30 min before stopping by adding 50 μl per well of 4 M H2SO4 solution (Thermo Scientific). Optical density was measured at 450 nm using a Microplate Reader (Synergy H1, Biotek). Data were analyzed using Prism 7 software (GraphPad), and values were reported as endpoint titers.
Competition ELISA
For the competition ELISA, the five highest inducers by cH6/1 ELISAs for each vaccination group were selected. Their sera were tested for antibodies competing with stalk-specific antibody CR9114 or HA trimer interface-binding antibody D1 H1-3/H3-3. Microtiter 96-well plates (Thermo Fisher) were coated with 50 µl of recombinant cH6/1 HA at a concentration of 2 µg ml−1 in PBS overnight at 4 °C. The following day, the plates were blocked with 220 µl of blocking buffer (PBS-T supplemented with 3% goat serum (Life Technologies) and 0.5% milk powder (AmericanBio)) for at least 1 h at 24 °C. Human serum samples were diluted to a starting concentration of 1:50 and serially diluted 1:2 in blocking solution. The plates were incubated for at least 2 h at room temperature and washed three times with PBS-T using an automatic plate washer (BioTek). Next, 100 µl per well of biotinylated monoclonal antibody CR9114 (at a concentration of 0.01 µg ml−1) or biotinylated antibody D1 H1-3/H3-3 (at a concentration of 0.5 µg ml−1), respectively, was added and the plates were incubated in the dark for 1 h at 24 °C. After 1 h, the plates were washed four times with PBS-T and 50 µl of streptavidin labeled with HRP (Thermo Fisher), diluted 1:3,000 in blocking solution, was added to each well and incubated for 1 h at 24 °C in the dark. The plates were washed four times with PBS-T and developed with SigmaFast OPD (Sigma) for 10 min, and the reaction was stopped with 3 M HCl (Thermo Fisher). The plates were read at a wavelength of 490 nm with a microplate reader (BioTek) and the data were analyzed in Microsoft Excel and GraphPad Prism 7. Percentage competition was calculated based on the average signal of antibody CR9114 only or D1 H1-3/H3-3 only, respectively, on each plate.
Microneutralization assay
Samples from d1, d28, d85, d113 and d420 were tested in a blinded fashion according to SOPs at ISMMS. Samples were treated with receptor-destroying enzyme (RDE; Denka-Seiken) for 18–20 h in a 37 °C water bath. Sodium citrate solution (2.5% w/v) was added and incubated for 1 h at 56 °C to stop RDE treatment. The RDE-treated serum samples were serially diluted twofold (starting dilution of 1:10) in UltraMDCK medium (Lonza), supplemented with tosyl phenylalanyl chloromethyl ketone-treated trypsin (infection medium; Sigma) at a concentration of 1 µg ml−1, in 96-well cell culture plates (Costar). The viruses (pH1N1, asH1N1, cH6/1N5 and H5N8) were diluted to concentrations of 100 × 50% cell culture infectious doses in infection medium. Serially diluted serum (60 µl) was incubated with 60 µl of diluted virus for 1 h at room temperature on a shaker. MDCK cells were washed with PBS, and 100 µl of the serum–virus mixture was added to the cells and incubated for 1 h at 33 °C. After 1 h, the serum–virus mixture was removed and replaced with diluted serum at the previous concentration. The plates were incubated at 33 °C for 48 h and the readout was performed by means of the classical hemagglutination assay. In brief, chicken red blood cells (RBCs; Lampire Biological Laboratories) were diluted to a concentration of 0.5% in PBS and added to 50 μl of cell supernatant in V-bottom 96-well plates (Corning). The plates were incubated at 4 °C for 45 min, scanned and the results analyzed in Microsoft Excel and visualized in GraphPad Prism 7.
K off measurement
Polyclonal serum Koff values were determined by biolayer interferometry using an Octet Red96 machine (ForteBio) with blinded serum samples from d1, d28, d85, d113 and d420. The serum samples were RDE treated overnight as described above to reduce nonspecific binding. The treated samples were further diluted in assay buffer consisting of PBS-T supplemented with 0.01% bosvine serum albumin (BSA; Sigma) to a starting concentration of 1:80. One Ni-NTA biosensor (ForteBio) per serum sample was soaked in assay buffer and a baseline was measured for 180 s. Recombinant HA (cH6/1) diluted to 10 µg ml−1 was loaded onto the sensors for 300 s. Next, a second baseline was measured for 120 s and each of the sensors was submerged into a different serum sample. Serum antibodies were allowed to associate with recombinant protein for 300 s and the sensors were submerged in buffer for 600 s for a dissociation step. All steps were performed at 26 °C and 1,000 r.p.m. shake speed. Longitudinal serum samples from the same subjects were measured at the same time and one additional sensor per subject was used to subtract the assay background. The data were analyzed using Octet Red96 operating software and a global curve fit was performed to determine the Koff rate.
Enzyme-linked lectin assay (ELLA)
The ELLA, used to determine NA inhibition activity, was performed as previously described in detail on blinded samples from d1, d28, d85, d113 and d420 (refs. 58,59). In brief, the neuraminidase activity of each virus was first determined in the absence of sera in an ELLA. The viruses (H6N3, H7N1) were diluted to 2 × half-maximal effective concentration and used in the subsequent NI assay. Serum samples were heat inactivated for 1 h at 56 °C and further diluted to a starting concentration of 1:20. The data were analyzed in Microsoft Excel and GraphPad Prism 7, and the half-maximum inhibitory concentration of each serum sample was calculated.
ADCC reporter assay
The ADCC reporter assay was performed on blinded samples from d1, d28, d85, d113 and d420 at ISMMS. In preparation for a series of repeated ADCC runs, target cells (MDCK and MDCK cH6/1 cell lines) were expanded using culture conditions described previously45 and a large batch of cryostocks was frozen and stored in liquid nitrogen. Once the target cells were put into culture, they were passaged for a maximum of 40 passages before the new cryostock was retrieved to avoid run-to-run variability due to prolonged culturing periods of the target cells. The effector cells (Jurkat cells expressing human FcɣRIIIa, high-affinity version V158, Promega) were expanded and frozen in a large batch in liquid nitrogen according to the manufacturer’s instructions (Promega). The recombinant monoclonal antibody CR9114 (ref. 60) was produced in large quantities in a single batch, aliquoted and stored at −20 °C. A fresh aliquot was used with every single run, diluted at 30 μg ml−1, as a maximal positive control, and diluted 1,000× (~1 μg ml−1) in each assay run as a secondary positive control; dilution was in Roswell Park Memorial Institute 1640 (RPMI 1640) medium (Gibco) spiked with human IgG-depleted serum. Human IgG-depleted serum (Innovative Research) was aliquoted into small aliquots and stored at −80 °C.
On day 0, the target cells were seeded at 7.5 × 104 cells per well into white, flat, 96-well plates (Corning) in cDMEM (Gibco) plus 10% FBS (HyClone) and cultivated at 37 °C and 5% CO2 overnight. On the same day, the effector cells were retrieved from the liquid nitrogen, counted and incubated overnight at 37 °C and 5% CO2. Human serum sample dilutions were prepared in U-shaped 96-well plates (Flacon) with initial dilution of 1:50, followed by threefold dilution series.
On day 1, the target cells were washed with 1× PBS (Gibco) and 25 μl of RPMI 1640 medium was pipetted into each well. Then, 25 μl of human serum samples and control monoclonal antibody dilutions were transferred from the dilution plate to the assay plate. The effector cells were counted again and 7.5 × 104 cells were added into each well in a total volume of 25 μl of RPMI medium. Assay plates were incubated for 6 h at 37 °C and 5% CO2. Following the 6-h incubation, the assay plates were removed from the cell culture incubator and equilibrated to room temperate for 15 min in a biosafety cabinet, before adding 75 μl of Bio-Glo luciferase substrate solution (Promega). After an additional incubation for 20 min at room temperature in the dark, the luminescence signal was measured using a BioTek Synergy H1 microplate reader (BioTek). The AUC baseline was calculated for each plate using the luminescence data obtained from the background control wells in Microsoft Excel. The AUC values were calculated using GraphPad Prism 7 (GraphPad) using the baseline values and reported.
ADCP reporter assay
The ADCP reporter assay was performed on blinded samples from d1, d28, d85, d113 and d420 at ISMMS. In preparation for serial ADCP runs, A549 cells used as target cells were expanded, frozen and stored in liquid nitrogen. The cH6/1N5 virus was grown as described above. The virus titer of the virus batch was determined using a plaque assay. The effector cells (Jurkat cells engineered to express human FcɣRIIa, high-affinity version H131, Promega) were expanded, frozen and stored in liquid nitrogen according to the manufacturer’s instructions (Promega). The same positive controls were employed as described above for the ADCC reporter assay. On day 0, A549 cells were seeded at 7.5 × 104 cells per well into white, flat, 96-well plates (Corning) in cDMEM medium and cultivated at 37 °C and 5% CO2 overnight. The effector cells were retrieved from the liquid nitrogen, counted and incubated overnight at 37 °C and 5% CO2. On day 1, the cDMEM medium was aspirated and target cells were washed once with PBS. They were then infected with the cH6/1N5 virus at a multiplicity of infection of 5, and resuspended in the fresh cDMEM medium (single cycle infection) in a total volume of 200 μl. The assay plate was then incubated at 37 °C and 5% CO2 for 16–24 h. The human serum dilutions were prepared the same way as described above for the ADCC reporter assay. On day 2, the assay set up, incubation and analysis were performed the same way as described above for the ADCC reporter assay.
Passive transfer mouse experiments
Serum pools for each vaccination group (negative control groups were combined; four separate groups) and visit (d1, d113, d420; three visits) were generated (12 serum pools in total). Only participants for whom serum was available for all three visits were included to allow for comparison within a group between visits. The pooling was performed by semi-unblinded staff in a laboratory not otherwise involved in the clinical trial. Each pool was assigned a numerical code, and study personnel remained blinded to visit and vaccine group. The serum transfer experiments were performed similarly as previously described40. Briefly, female, 6–8-week-old BALB/c mice were randomly assigned to each serum pool and each mouse was administered 150 μl of serum intraperitoneally. At 2–6 h after serum transfer, mice were sedated by ketamine/xylazine anesthesia, blood was collected to confirm successful serum transfer and mice were intranasally challenged with virus diluted in 50 μl of PBS. To measure the HA stalk-specific protective effect of the serum samples, mice were challenged using cH6/1N5 virus (described above), which expresses an avian HA head domain and NA (to which humans are generally naïve), but the conserved H1 stalk domain. To assess survival and weight loss, mice (n = 10 per group per challenge virus per study visit) were weighed daily and monitored for severe symptoms after challenge. Mice that lost more than 25% of their initial body weight were euthanized. For lung titer measurements, mice were euthanized on either day 3 (n = 5 per group per study visit) or day 6 (n = 5 per group per study visit) after challenge. Their lungs were collected and homogenized, supernatants were clarified by centrifugation and aliquots were frozen before further testing by plaque assay. The plaque assays were performed as previously described61
HI assay
Sera were assessed for antibody to various influenza virus isolates or chimeric strains to determine antibody reaction after vaccination to the HA by HI assay using standard methods62,63. In brief, sera were treated with RDE (Denka-Seiken) to remove nonspecific inhibitors of hemagglutination before testing. Following RDE treatment, the samples were further diluted to 1:10 in PBS. The sera were then treated with packed RBCs to remove nonspecific agglutinins. The RBCs were spun out of the sera and the samples were ready for testing. Starting at a 1:10 dilution, the sera were diluted twofold through 1:2,560 in V-bottom microtiter plates.
The inactivated viral antigens (cH6/1N5, cH8/1N1, cH5/1N1, pandemic H1N1, avian-swine H1N1 and H5N8—all described above) were added to serially diluted sera, and incubated at room temperature for 30 min. Turkey RBCs (Viromed Laboratories) were suspended at a concentration of 0.5% in PBS, added to the serum–viral antigen mixture and incubated at room temperature for 30 min. Plates were tilted and read. The antibody titers were reported as the reciprocal of the last serum dilution to completely inhibit RBC agglutination. Sera without reactivity were assigned a value of <10. Sera with initial titers of ≥2,560 were retested at a higher starting dilution to obtain a reportable titer. Control sera were established for each antigen and were run in each assay. The assay was valid if the control sera fell within twofold of their defined titer.
Detection of influenza virus by real-time RT–PCR
Nasal swabs collected from the subjects were tested for the presence of influenza virus by RT–PCR. Nucleic acid extraction of 140 µl of the swab samples was carried out by use of the Qiagen QIAamp Viral RNA Mini Kit (Qiagen). Primers and probes (Biosearch Technologies) targeting the HA gene of the pandemic (pdmH1) influenza A (H1N1) 2009 virus were used and are listed in Supplementary Table 3. To evaluate the quality of the swab samples, a separate PCR reaction was performed to detect the Human RNPase P gene. Detection of this gene confirms that the swab sample is of sufficient quality that cell-associated virus can be detected and quantified and acts as an internal control for any possible PCR inhibitors in the swab sample. A one-step quantitative reverse transcriptase–probe hydrolysis kit, Ambion AgPath-ID One-Step kit (Thermo Fisher), was used in the PCR reaction following manufacturer’s instructions. Final concentration of primers was 0.8 µM and 0.2 µM for the probe. For each reaction, 5 µl of the extracted material was used. PCR conditions using an Applied Biosystems ABI 7500 PCR system (Thermo Fisher) were as follows: 50.0 °C for 30 min; 95.0 °C for 10 min; 45 cycles of 95 °C, 15 s followed by 55 °C for 34 s. Swab specimens that were positive for H1N1 were then tested by RT–PCR using primer-probe sets designed to specifically detect the cH8/1 HA gene. The primer-probe set was shown to detect cH8/1N1 and not H1N1 viruses. RT–PCR conditions were the same as above and the primers and probes are listed in Supplementary Table 3.
Growth and determination of titer of influenza viruses
To determine if live virus could be recovered from the swab samples that tested positive by RT–PCR, MDCK cells in culture tubes in Zero-Serum Media (Diagnostic Hybrids) with 4 μg ml−1 trypsin (trypsin 1:250, Gibco BRL) were infected with 200 µl of swab sample. Daily cytopathic effect was noted. After 2 d of incubation, immunostaining was performed. The cell layer was removed by scraping, suspended in PBS and fixed to a chamber slide. To detect the presence of the cH8/1N1 virus, a monoclonal antibody (1A7) specific to the virus was used52. The cells were incubated with the monoclonal antibody and then with a secondary fluorescein-conjugated goat anti-mouse IgG (Chemicon International) antibody. The presence of fluorescence in cells stained with the specific monoclonal antibody was determined.
Statistical analysis
Sample size was determined primarily based on safety considerations, but power calculations determined that it would permit detection of GMT ratios ≥3 of anti-H1 stalk humoral and mucosal immune responses (serum/salivary IgG, IgA, serum neutralizing antibody and ADCC activity) between actively immunized groups with power ≥86% assuming a standard deviation of log10 titers ≤0.4 with ≥14 evaluable subjects retained per actively immunized group, allowing for a limited amount of dropout from the number enrolled, using two-sided type I error of 0.05. Additional subjects were enrolled in group 1 to ensure adequate power for comparisons involving this high-priority group if dropout was higher than anticipated29. Immunogenicity analyses were conducted primarily in the per-protocol population, which included all participants without major deviations, including those considered likely to affect the immune response, such as participation in other clinical studies or a positive test for influenza virus during the course of the study. All analyses are pooled across study sites. No adjustments were made for multiplicity of testing due to the exploratory nature of many of the outcomes, the large number of tests performed and the modest sample size aimed at a preliminary evaluation. Statistical analysis was done with SAS software v.9.4.
Safety
Primary endpoints of safety and tolerability were the number and percentage of subjects experiencing at least one AE, and the number and percentage of subjects experiencing each specific AE, tabulated by study group (for individual postdose periods and for the entire study period) and product administered (for individual postdose periods) along with their corresponding exact 95% confidence intervals (CIs). Safety results were summarized among all subjects receiving at least one vaccine, according to vaccine received.
Solicited AEs were summarized overall by study group and product received and by type, as well as by severity grade. Immediate solicited AEs recorded by study staff, as well as a combined analysis of immediate and subject-reported events within 7 d, were summarized according to the number and proportion of subjects reporting at least one event of any type, any systemic event or any local event. The 95% CIs were calculated using Clopper–Pearson methodology, and a two-sided Fisher’s exact test was used as a global test for a difference among groups, grouping by study product where possible.
Unsolicited AEs reported by the individual or detected by study personnel were recorded and were coded with the Medical Dictionary for Regulatory Activities system organ class and preferred term. For analysis, each subject was only counted once per category and any repetition of AEs within a subject was ignored. Per protocol, all AEs occurring within 28 d of a vaccination were presented. SAEs, pIMDs, medically attended events and laboratory-confirmed ILIs occurring at any point were also presented. Events were summarized at the subject level by group and study product (where relevant) overall and by type, severity grade and according to investigator determination of relationship to study product under the maximum severity and/or relationship for each event type, as appropriate.
Immunogenicity
For immunogenicity endpoints, descriptive statistics (for example, mean, standard deviation, median, range) were tabulated by study group and time point, based on the log transformation where relevant, along with the GMT and its 95% CI, where relevant. The two-sided 95% CI for each GMT was obtained using a t-distribution on log-transformed titers/concentrations. Additionally, the geometric mean fold rise from baseline and accompanying t-distribution-based two-sided 95% CI were computed for relevant immunogenicity endpoints, where the fold rise at postbaseline visit for each subject was computed as the antilog of the difference in log titer of postbaseline measurement minus baseline. CIs for the median were computed via the percentile bootstrap on the log scale, with n = 10,000 replicates, then back-transformed. Categorical outcomes (for example, seroresponse rates (≥4-fold or ≥10-fold rise after vaccination), post-LAIV viral shedding) were summarized as the proportion, along with corresponding two-sided exact 95% CI.
Wherever assay results were less than the lower limit of quantitation (LLOQ), one-half of the LLOQ was used for computation of means and standard deviations on the log scale. For percentiles and any accompanying CIs, values not achieving the LLOQ were presented as ‘<LLOQ’ with LLOQ replaced by the relevant numeric quantity. For all analyses involving parametric methods based on the t-distribution, if substantial non-normality and/or values achieving the LLOQ were frequent, a corresponding nonparametric method was to be substituted.
The mouse passive transfer study targeted evaluation of differences in protection from challenge by transfer of serum obtained pre- and postvaccination within groups, as well as postvaccination between groups. The percentage weight loss was summarized descriptively, with mean percentage weight loss by postchallenge day augmented with 95% percentile bootstrap-based CIs. Mice that were excluded or reached the humane endpoint before a given study day did not contribute to the computation at subsequent study days.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The protocol, statistical analysis plan and results will be available at https://clinicaltrials.gov/ct2/show/NCT03300050. Underlying data are available upon request (excluding identifiable information).
References
Palese, P. Influenza: old and new threats. Nat. Med. 10, S82–S87 (2004).
Chan, M. C. W. et al. Frequent genetic mismatch between vaccine strains and circulating seasonal influenza viruses, Hong Kong, China, 1996–2012. Emerg. Infect. Dis. 24, 1825–1834 (2018).
Flannery, B. et al. Spread of antigenically drifted influenza A(H3N2) viruses and vaccine effectiveness in the United States during the 2018–2019 season. J. Infect. Dis. 221, 8–15 (2020).
Skowronski, D. M. et al. Paradoxical clade- and age-specific vaccine effectiveness during the 2018/19 influenza A(H3N2) epidemic in Canada: potential imprint-regulated effect of vaccine (I-REV). Euro Surveill. 24, 1900585 (2019).
Owusu, D. et al. Early season pediatric influenza B/Victoria virus infections associated with a recently emerged virus subclade—Louisiana, 2019. MMWR Morb. Mortal. Wkly Rep. 69, 40–43 (2020).
Virk, R. K. et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl Acad. Sci. USA 117, 619–628 (2020).
Krammer, F. & Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 14, 167–182 (2015).
Krammer, F. The human antibody response to influenza A virus infection and vaccination. Nat. Rev. Immunol. 19, 383–397 (2019).
Hobson, D., Curry, R. L., Beare, A. S. & Ward-Gardner, A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J. Hyg. (Lond.) 70, 767–777 (1972).
Heaton, N. S., Sachs, D., Chen, C. J., Hai, R. & Palese, P. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc. Natl Acad. Sci. USA 110, 20248–20253 (2013).
Doud, M. B. & Bloom, J. D. Accurate measurement of the effects of all amino-acid mutations on influenza hemagglutinin. Viruses 8, 155 (2016).
Kirkpatrick, E., Qiu, X., Wilson, P. C., Bahl, J. & Krammer, F. The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci. Rep. 8, 10432 (2018).
Okuno, Y., Isegawa, Y., Sasao, F. & Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 67, 2552–2558 (1993).
Wu, N. C. & Wilson, I. A. Structural insights into the design of novel anti-influenza therapies. Nat. Struct. Mol. Biol. 25, 115–121 (2018).
Corti, D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011).
Dreyfus, C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012).
Ng, S. et al. Novel correlates of protection against pandemic H1N1 influenza A virus infection. Nat. Med. 25, 962–967 (2019).
Dilillo, D. J., Tan, G. S., Palese, P. & Ravetch, J. V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 20, 143–151 (2014).
Brandenburg, B. et al. Mechanisms of hemagglutinin targeted influenza virus neutralization. PLoS ONE 8, e80034 (2013).
Rajendran, M. et al. Analysis of anti-influenza virus neuraminidase antibodies in children, adults, and the elderly by ELISA and enzyme inhibition: evidence for original antigenic Sin. mBio 8, e02281–16 (2017).
Hai, R. et al. Influenza viruses expressing chimeric hemagglutinins: globular head and stalk domains derived from different subtypes. J. Virol. 86, 5774–5781 (2012).
Margine, I. et al. Hemagglutinin stalk-based universal vaccine constructs protect against group 2 influenza A viruses. J. Virol. 87, 10435–10446 (2013).
Krammer, F., Pica, N., Hai, R., Margine, I. & Palese, P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J. Virol. 87, 6542–6550 (2013).
Nachbagauer, R. et al. A universal influenza virus vaccine candidate confers protection against pandemic H1N1 infection in preclinical ferret studies. NPJ Vaccines 2, 26 (2017).
Choi, A. et al. Chimeric hemagglutinin-based influenza virus vaccines induce protective stalk-specific humoral immunity and cellular responses in mice. Immunohorizons 3, 133–148 (2019).
Nachbagauer, R. et al. A chimeric haemagglutinin-based influenza split virion vaccine adjuvanted with AS03 induces protective stalk-reactive antibodies in mice. NPJ Vaccines 1, 16015 (2016).
Nachbagauer, R. et al. Age dependence and isotype specificity of influenza virus hemagglutinin stalk-reactive antibodies in humans. mBio 7, e01996–15 (2016).
Sui, J. et al. Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza A antibodies. Clin. Infect. Dis. 52, 1003–1009 (2011).
Bernstein, D. I. et al. Immunogenicity of chimeric haemagglutinin-based, universal influenza virus vaccine candidates: interim results of a randomised, placebo-controlled, phase 1 clinical trial. Lancet Infect. Dis. 20, 80–91 (2020).
Talaat, K. R. et al. A live attenuated influenza A(H5N1) vaccine induces long-term immunity in the absence of a primary antibody response. J. Infect. Dis. 209, 1860–1869 (2014).
Rudenko, L. et al. Assessment of immune responses to H5N1 inactivated influenza vaccine among individuals previously primed with H5N2 live attenuated influenza vaccine. Hum. Vaccin. Immunother. 11, 2839–2848 (2015).
Barría, M. I. et al. Localized mucosal response to intranasal live attenuated influenza vaccine in adults. J. Infect. Dis. 207, 115–124 (2013).
Lee, J. et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat. Med. 22, 1456–1464 (2016).
Ekiert, D. C. et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489, 526–532 (2012).
Khurana, S. et al. MF59 adjuvant enhances diversity and affinity of antibody-mediated immune response to pandemic influenza vaccines. Sci. Transl. Med. 3, 85ra48 (2011).
Ekiert, D. C. et al. Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251 (2009).
Sui, J. et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273 (2009).
Nachbagauer, R. et al. Induction of broadly reactive anti-hemagglutinin stalk antibodies by an H5N1 vaccine in humans. J. Virol. 88, 13260–13268 (2014).
Ellebedy, A. H. et al. Induction of broadly cross-reactive antibody responses to the influenza HA stem region following H5N1 vaccination in humans. Proc. Natl Acad. Sci. USA 111, 13133–13138 (2014).
Jacobsen, H. et al. Influenza virus hemagglutinin stalk-specific antibodies in human serum are a surrogate marker for in vivo protection in a serum transfer mouse challenge model. mBio 8, e01463–17 (2017).
Bangaru, S. et al. A site of vulnerability on the influenza virus hemagglutinin head domain trimer interface. Cell 177, 1136–1152.e1118 (2019).
Watanabe, A. et al. Antibodies to a conserved influenza head interface epitope protect by an IgG subtype-dependent mechanism. Cell 177, 1124–1135.e1116 (2019).
Nachbagauer, R. et al. Pandemic influenza virus vaccines boost hemagglutinin stalk-specific antibody responses in primed adult and pediatric cohorts. NPJ Vaccines 4, 51 (2019).
He, W. et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 8, 846 (2017).
Chromikova, V. et al. Activity of human serum antibodies in an influenza virus hemagglutinin stalk-based ADCC reporter assay correlates with activity in a CD107a degranulation assay. Vaccine 38, 1953–1961 (2020).
Cohet, C. et al. Safety of AS03-adjuvanted influenza vaccines: a review of the evidence. Vaccine 37, 3006–3021 (2019).
Krammer, F. et al. H3 stalk-based chimeric hemagglutinin influenza virus constructs protect mice from H7N9 challenge. J. Virol. 88, 2340–2343 (2014).
Sun, W. et al. Development of influenza B universal vaccine candidates using the ‘mosaic’ hemagglutinin approach. J. Virol. 93, e00333–19 (2019).
Pitisuttithum, P. et al. Safety and immunogenicity of a live attenuated influenza H5 candidate vaccine strain A/17/turkey/Turkey/05/133 H5N2 and its priming effects for potential pre-pandemic use: a randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 17, 833–842 (2017).
Isakova-Sivak, I. et al. Genetic bases of the temperature-sensitive phenotype of a master donor virus used in live attenuated influenza vaccines: A/Leningrad/134/17/57 (H2N2). Virology 412, 297–305 (2011).
Isakova-Sivak, I. et al. Broadly protective anti-hemagglutinin stalk antibodies induced by live attenuated influenza vaccine expressing chimeric hemagglutinin. Virology 518, 313–323 (2018).
Rajendran, M. et al. An immuno-assay to quantify influenza virus hemagglutinin with correctly folded stalk domains in vaccine preparations. PLoS ONE 13, e0194830 (2018).
Martínez-Sobrido, L. & García-Sastre, A. Generation of recombinant influenza virus from plasmid DNA. J. Vis. Exp. 3, 2057 (2010).
Hong, M. et al. Antibody recognition of the pandemic H1N1 influenza virus hemagglutinin receptor binding site. J. Virol. 87, 12471–12480 (2013).
Margine, I., Palese, P. & Krammer, F. Expression of functional recombinant hemagglutinin and neuraminidase proteins from the novel H7N9 influenza virus using the baculovirus expression system. J. Vis. Exp. 6, e51112 (2013).
Impagliazzo, A. et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 349, 1301–1306 (2015).
Bliss, C. M. et al. Targeting antigen to the surface of EVs improves the in vivo immunogenicity of human and non-human adenoviral vaccines in mice. Mol. Ther. Methods Clin. Dev. 16, 108–125 (2020).
Stadlbauer, D. et al. Broadly protective human antibodies that target the active site of influenza virus neuraminidase. Science 366, 499–504 (2019).
Wang, G. et al. Characterization of swine-origin H1N1 canine influenza viruses. Emerg. Microbes Infect. 8, 1017–1026 (2019).
Chromikova, V., Zaragoza, M. A. & Krammer, F. Generation of a serum free CHO DG44 cell line stably producing a broadly protective anti-influenza virus monoclonal antibody. PLoS ONE 12, e0183315 (2017).
McMahon, M. et al. Vaccination with viral vectors expressing chimeric hemagglutinin, NP and M1 antigens protects ferrets against influenza virus challenge. Front. Immunol. 10, 2005 (2019).
Lu, Y. et al. Immune response to influenza vaccine in children with inflammatory bowel disease. Am. J. Gastroenterol. 104, 444–453 (2009).
Treanor, J. et al. Safety and immunogenicity of a baculovirus-expressed hemagglutinin influenza vaccine: a randomized controlled trial. JAMA 297, 1577–1582 (2007).
Acknowledgements
We thank the research team at the Department of Microbiology at the Icahn School of Medicine at Mount Sinai for supporting the development of this vaccine concept since 2011. We also thank the teams at PATH (with special acknowledgement to J.C. Victor, R. Wahid and K. Mahmood), Cincinnati Children’s Hospital Medical Center, Duke University, the University of Chicago, GSK, NEOMED-LABS and the EMMES Corporation for their work on this project. We are grateful for help from V. Simon at Mount Sinai with IRB protocol development, D. Wentworth and L. M. Chen at the US CDC with the initial LAIV seed virus rescue and for help from D. Suarez from the US Department of Agriculture for pathogenicity testing of the vaccine virus strains. We thank G. Georgiou for providing monoclonal antibody D1 H1-3/H3-3 used in competition ELISAs. This study was funded in part by the Bill and Melinda Gates Foundation (grant OPP1084518). Basic research leading to this trial was supported by the National Institute of Allergy and Infectious Diseases (NIAID) (grants P01 AI097092, HHSN26620070010C, HHSN272201400008C, U19 AI109946, R01 AI128821). NIAID continues to support analysis of samples from this trial through a Collaborative Influenza Vaccine Innovation Centers contract (75N93019C00051). GSK made adjuvants and vaccine preparations available and contributed through providing expertise as well (funding for this study was provided by GlaxoSmithKline Biologicals SA). The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Gates Foundation. GlaxoSmithKline Biologicals SA was provided the opportunity to review a preliminary version of this manuscript for factual accuracy, but the authors are solely responsible for final content and interpretation.
Author information
Authors and Affiliations
Contributions
J.F., A.N., D.I.B., J.G., E.B.W., F.B.-S., C. Claeys, C.G., C.M., M.M.N., A.S., M.V.d.W. and B.L.I. are responsible for conducting, managing and coordinating the clinical trial. Laboratory experiments were performed by R.N., D.S., T.A., M.A.B., D.B., C.B., C. Capuano, J.M.C., V.C., L.C., A.W.F., A.J., K.J., M.M.M., M.M.N., S.S. and W.S. Resources and methods were provided by L.C., T.A., W.S., A.W.F., V.C., S.S., P.C.W. and F.K. All authors are responsible for writing, reviewing and editing the manuscript. Data analysis was performed by R.N., J.F., A.N., F.B.-S., D.S., J.M.C., V.C., C.G., C.M., A.S., B.L.I., A.G.-S., P.P. and F.K. Quality assurance on assays was performed at ISMMS by C.M.
Corresponding author
Ethics declarations
Competing interests
The Icahn School of Medicine at Mount Sinai (ISMMS) has issued patents and filed patent applications covering the use of chimeric hemagglutinin antigens as vaccines. R.N., A.G.-S., P.P. and F.K. are named as inventors on these patents and applications. The ISMMS and the inventors have received payments as consideration for these rights. The laboratories of A.G.-S., P.P. and F.K. were also engaged in a research program that was funded by GlaxoSmithKline Biologicals SA. M.V.d.W. declares that she is employed by the GSK group of companies and declares no nonfinancial conflicts of interest. C. Claeys was an employee of the GSK group of companies at the time the study was performed. B.L.I. was an employee of the GSK group of companies until 2017 and held shares from this group of companies at the time the study was performed. He is also named as inventor on a patent family regarding influenza virus vaccine constructs filed by the ISMMS and the GSK group of companies. E.B.W. is a coinvestigator for a clinical trial supported by Pfizer. All other authors declare no competing interests.
Additional information
Peer review information Alison Farrell is the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Microneutralization titers.
Microneutralization titers against cH6/1N5 virus (a), pandemic H1N1 virus (c), avian-swine H1N1 virus (e) and H5N8 virus (g). Faint lines indicate reactivity of different individuals; bold lines indicate geometric mean titers of the respective groups. Biologically independent samples; group 1: n = 19, group 2: n = 14, group 4: n = 15 groups 3 + 5: n = 13(3 + 10); examined in one independent experiment. Geometric mean fold induction of antibody titers based on data in a, c, e and g is shown in b, d, f and h.
Extended Data Fig. 2 Hemagglutination inhibition (HI) titers.
Hemagglutination inhibition titers against cH6/1N5 virus (a), pandemic H1N1 virus (c), avian-swine H1N1 virus (e) and H5N8 virus (g) as well as cH8/1N1 (I) and cH5/1N1 (k) vaccine strains. Faint lines indicate reactivity of different individuals; bold lines indicate geometric mean titers of the respective groups. Biologically independent samples; group 1: n = 19, group 2: n = 14, group 4: n = 15 groups 3 + 5: n = 13(3 + 10); examined in one independent experiment. Geometric mean fold induction of antibody titers based on data in a, c, e, g, i and k is shown in b, d, f, h, j and l.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and Supplementary Tables 1–3.
Rights and permissions
About this article

Cite this article
Nachbagauer, R., Feser, J., Naficy, A. et al. A chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase I trial. Nat Med 27, 106–114 (2021). https://doi.org/10.1038/s41591-020-1118-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-1118-7
This article is cited by
-
Scientific highlights of the 9th ESWI Influenza Conference
One Health Outlook (2024)
-
Absolute quantitation of binding antibodies from clinical samples
npj Vaccines (2024)
-
Vaccine design via antigen reorientation
Nature Chemical Biology (2024)
-
Bringing immunofocusing into focus
npj Vaccines (2024)
-
Hemagglutinin virus-like particles incorporated with membrane-bound cytokine adjuvants provide protection against homologous and heterologous influenza virus challenge in aged mice
Immunity & Ageing (2023)
