
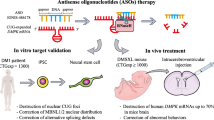
Abstract
CLN3 Batten disease is an autosomal recessive, neurodegenerative, lysosomal storage disease caused by mutations in CLN3, which encodes a lysosomal membrane protein1,2,3. There are no disease-modifying treatments for this disease that affects up to 1 in 25,000 births, has an onset of symptoms in early childhood and typically is fatal by 20–30 years of life4,5,6,7. Most patients with CLN3 Batten have a deletion encompassing exons 7 and 8 (CLN3∆ex7/8), creating a reading frameshift7,8. Here we demonstrate that mice with this deletion can be effectively treated using an antisense oligonucleotide (ASO) that induces exon skipping to restore the open reading frame. A single treatment of neonatal mice with an exon 5-targeted ASO-induced robust exon skipping for more than a year, improved motor coordination, reduced histopathology in Cln3∆ex7/8 mice and increased survival in a new mouse model of the disease. ASOs also induced exon skipping in cell lines derived from patients with CLN3 Batten disease. Our findings demonstrate the utility of ASO-based reading-frame correction as an approach to treat CLN3 Batten disease and broaden the therapeutic landscape for ASOs in the treatment of other diseases using a similar strategy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its extended data and supplementary information files. Source data are provided with this paper.
References
Kyttala, A., Ihrke, G., Vesa, J., Schell, M. J. & Luzio, J. P. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol. Biol. Cell 15, 1313–1323 (2004).
The International Batten Disease Consortium Isolation of a novel gene underlying Batten disease, CLN3. Cell 82, 949–957 (1995).
Cárcel-Trullols, J., Kovács, A. D. & Pearce, D. A. Cell biology of the NCL proteins: what they do and don’t do. Biochim. Biophys. Acta 1852, 2242–2255 (2015).
Cialone, J. et al. Females experience a more severe disease course in Batten disease. J. Inherit. Metab. Dis. 35, 549–555 (2012).
Gardiner, R. M. Clinical features and molecular genetic basis of the neuronal ceroid lipofuscinoses. Adv. Neurol. 89, 211–215 (2002).
Johnson, T. B. et al. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat. Rev. Neurol. 15, 161–178 (2019).
Sleat, D. E., Gedvilaite, E., Zhang, Y., Lobel, P. & Xing, J. Analysis of large-scale whole exome sequencing data to determine the prevalence of genetically-distinct forms of neuronal ceroid lipofuscinosis. Gene 593, 284–291 (2016).
Mole, S. E. & Cotman, S. L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta 1852, 2237–2241 (2015).
Kousi, M., Lehesjoki, A. E. & Mole, S. E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 33, 42–63 (2012).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345 (2014).
Cao, Y. et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 281, 20483–20493 (2006).
Chang, J. W., Choi, H., Cotman, S. L. & Jung, Y. K. Lithium rescues the impaired autophagy process in CbCln3(Δex7/8/Δex7/8) cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition. J. Neurochem. 116, 659–668 (2011).
Chandrachud, U. et al. Unbiased cell-based screening in a neuronal cell model of Batten disease highlights an interaction between Ca2+ homeostasis, autophagy, and CLN3 protein function. J. Biol. Chem. 290, 14361–14380 (2015).
Vidal-Donet, J. M., Cárcel-Trullols, J., Casanova, B., Aguado, C. & Knecht, E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One 8, e55526 (2013).
Yasa, S. et al. CLN3 regulates endosomal function by modulating Rab7A-effector interactions. J. Cell Sci. 133, jcs234047 (2020).
Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016).
Havens, M. A. & Hastings, M. L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 44, 6549–6563 (2016).
Cotman, S. L. et al. Cln3(Δex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 11, 2709–2721 (2002).
Tyynela, J., Cooper, J. D., Khan, M. N., Shemilts, S. J. & Haltia, M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. 14, 349–357 (2004).
Osorio, N. S. et al. Neurodevelopmental delay in the Cln3Δex7/8 mouse model for Batten disease. Genes Brain Behav. 8, 337–345 (2009).
Pontikis, C. C. et al. Late onset neurodegeneration in the Cln3 -/- mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 1023, 231–242 (2004).
Burkovetskaya, M. et al. Evidence for aberrant astrocyte hemichannel activity in juvenile neuronal ceroid lipofuscinosis (JNCL). PLoS One 9, e95023 (2014).
Sondhi, D. et al. Partial correction of the CNS lysosomal storage defect in a mouse model of juvenile neuronal ceroid lipofuscinosis by neonatal CNS administration of an adeno-associated virus serotype rh.10 vector expressing the human CLN3 gene. Hum. Gene Ther. 25, 223–239 (2014).
Aldrich, A. et al. Efficacy of phosphodiesterase-4 inhibitors in juvenile Batten disease (CLN3). Ann. Neurol. 80, 909–923 (2016).
Kovács, A. D. & Pearce, D. A. Finding the most appropriate mouse model of juvenile CLN3 (Batten) disease for therapeutic studies: the importance of genetic background and gender. Dis. Model Mech. 8, 351–361 (2015).
Wang, X., Huang, T., Bu, G. & Xu, H. Dysregulation of protein trafficking in neurodegeneration. Mol. Neurodegener. 9, 31 (2014).
Yang, D. S. et al. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain 137, 3300–3318 (2014).
Nilsson, P. et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69 (2013).
Wisniewski, K. E., Kida, E., Gordon-Majszak, W. & Saitoh, T. Altered amyloid β-protein precursor processing in brains of patients with neuronal ceroid lipofuscinosis. Neurosci. Lett. 120, 94–96 (1990).
Wisniewski, K. E., Gordon-Krajcer, W. & Kida, E. Abnormal processing of carboxy-terminal fragment of beta precursor protein (βPP) in neuronal ceroid-lipofuscinosis (NCL) cases. J. Inherit. Metab. Dis. 16, 312–316 (1993).
D’Andrea, M. R. et al. Lipofuscin and Aβ42 exhibit distinct distribution patterns in normal and Alzheimer’s disease brains. Neurosci. Lett. 323, 45–49 (2002).
Maulik, M. et al. Mutant human APP exacerbates pathology in a mouse model of NPC and its reversal by a β-cyclodextrin. Hum. Mol. Genet. 21, 4857–4875 (2012).
Chishti, M. A. et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 (2001).
Darras, B. T. et al. An integrated safety analysis of infants and children with symptomatic spinal muscular atrophy (SMA) treated with Nnusinersen in seven clinical trials. CNS Drugs 33, 919–932 (2019).
McClorey, G. & Wood, M. J. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr. Opin. Pharm. 24, 52–58 (2015).
Foust, K. D. et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274 (2010).
Dangouloff, T. & Servais, L. Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther. Clin. Risk Manag. 15, 1153–1161 (2019).
De Vivo, D. C. et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the Phase 2 NURTURE study. Neuromuscul. Disord. 29, 842–856 (2019).
Ke, Q. et al. Progress in treatment and newborn screening for Duchenne muscular dystrophy and spinal muscular atrophy. World J. Pediatr. 15, 219–225 (2019).
Viklund, H. & Elofsson, A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668 (2008).
Reynolds, S. M., Kall, L., Riffle, M. E., Bilmes, J. A. & Noble, W. S. Transmembrane topology and signal peptide prediction using dynamic bayesian networks. PLoS Comput. Biol. 4, e1000213 (2008).
Kall, L., Krogh, A. & Sonnhammer, E. L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 (2004).
Kall, L., Krogh, A. & Sonnhammer, E. L. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 21, i251–i257 (2005).
Bernsel, A. et al. Prediction of membrane-protein topology from first principles. Proc. Natl Acad. Sci. USA 105, 7177–7181 (2008).
Viklund, H., Bernsel, A., Skwark, M. & Elofsson, A. SPOCTOPUS: a combined predictor of signal peptides and membrane protein topology. Bioinformatics 24, 2928–2929 (2008).
Tsirigos, K. D., Peters, C., Shu, N., Kall, L. & Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 43, W401–W407 (2015).
Nugent, T., Mole, S. E. & Jones, D. T. The transmembrane topology of Batten disease protein CLN3 determined by consensus computational prediction constrained by experimental data. FEBS Lett. 582, 1019–1024 (2008).
Perland, E. et al. Characteristics of 29 novel atypical solute carriers of major facilitator superfamily type: evolutionary conservation, predicted structure and neuronal co-expression. Open Biol. 7, 170142 (2017).
Yang, J. & Zhang, Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 (2015).
Roy, A., Kucukural, A. & Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010).
Omasits, U., Ahrens, C. H., Muller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 (2014).
Chresta, C. M. et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298 (2010).
Swayze, E. E. et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 35, 687–700 (2007).
Rigo, F. et al. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharm. Exp. Ther. 350, 46–55 (2014).
Baker, B. F. et al. 2′-O-(2-Methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J. Biol. Chem. 272, 11994–12000 (1997).
Hinrich, A. J. et al. Therapeutic correction of ApoER2 splicing in Alzheimer’s disease mice using antisense oligonucleotides. EMBO Mol. Med. 8, 328–345 (2016).
Hua, Y. & Krainer, A. R. Antisense-mediated exon inclusion. Methods Mol. Biol. 867, 307–323 (2012).
Kovács, A. D. et al. Temporary inhibition of AMPA receptors induces a prolonged improvement of motor performance in a mouse model of juvenile Batten disease. Neuropharmacology 60, 405–409 (2011).
Acknowledgements
The authors thank Forbes Porter and An Dang Do (NIH/NICHD) and Beverly Davidson (Children’s Hospital of Philadelphia) for human fibroblast cell lines and David Pearce (Sanford), David Mueller (RFUMS) and Susan Cotman (Harvard) for helpful discussions and other reagents. The authors also thank Maria Ruiz and Cecilia Reyes for technical support. This work was supported by NIH grant NS113233, the Batten Disease Support and Research Alliance and the ForeBatten Research Foundation. Quantitation of radioactive PCR products was performed on an instrument in the shared Proteomics facility at RFUMS obtained with NIH grant S10 OD010662.
Author information
Authors and Affiliations
Contributions
J.L.C., F.M.J. and M.L.H. designed and carried out mouse behavioral analysis; J.L.C., A.J.H. and M.L.H. designed and performed in vitro ASO experiments; J.L.C. and A.J.H. performed immunofluorescent analysis of ASO distribution; T.B.J and J.M.W. carried out immunohistochemistry for SCMAS accumulation and GFAP; J.O. and F.R. designed, performed and analyzed data from experiments to measure autophagy and lysosomal volume; M.J. and F.R. designed and performed adult ICV treatments. J.M.W., F.R., D.M.D and M.L.H. provided critical materials and reagents. J.L.C. and M.L.H. wrote the manuscript. All authors discussed the results and contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.O, M.J. and F.R. are employees of Ionis Pharmaceuticals. T.B.J. and J.M.W. are currently employees of Amicus Therapeutics. D.M.D. is currently employed by Abbott Molecular. M.L.H. and F.R. are inventors on patents on ASOs filed by RFUMS and Ionis Pharmaceuticals and may be entitled to benefits from licensing of the associated intellectual property.
Additional information
Peer review information Kate Gao was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Modeling of CLN3 protein structure.
a, A query of the structures of CLN3 using different prediction algorithms, generated models of human CLN3. The table shows the protein prediction servers with their predicted number of transmembrane domains (TMDs) for CLN3 (WT), CLN3Δex7/8 (Δ78), and CLN3Δex5/7/8 (Δ578). The variability of TMDs amongst the different predictions demonstrates the uncertainty associated with such modeling approaches and cautions that definitive conclusions about CLN3 structures must await high resolution information on the structure from cryo-EM or X-ray crystallographic studies. b, Homology model of the CLN3 structure based on the structure of the equilibrative nucleoside transporter (ENT1) encoded by SLC29A1, as a template, generated by I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). c, Membrane topology map of CLN3, CLN3Δex7/8 (Δ78), and CLN3Δex5/7/8 (Δ578) showing the numbered TMDs as predicted by TOPCONS and rendered using Protter (http://wlab.ethz.ch/protter). The exons corresponding to the amino acid sequence are shown in alternating green and white with exon 5-encoded amino acids highlighted in blue.
Extended Data Fig. 2 Autophagic flux deficits associated with CLN3Δex7/8 but not CLN3Δex5/7/8 expression.
a, Schematic representation of measuring autophagic flux using GFP-RFP-LC3B. To compare the effects of CLN3 protein isoforms on autophagic flux we used a tandem acid-stable red and acid-labile green fluorescent protein (RFP, GFP) sensor fused to the autophagosome-associated protein, microtubule-associated protein light chain 3 beta (GFP-RFP-LC3B) in HEK-293 cells. The GFP signal is quenched in acidic condition and thereby the dual-labeled RFP-GFP-LC3B provides a means to differentiate between autophagosomes (red and green=yellow fluorescent LC3B) and acidic autolysosomes (red only) and assess the relative abundance of the vesicles during autophagy. LC3BII localizes to the membrane of the autophagosome, which fuses with lysosomes to form acidic autolysosomes. This acidic environment causes quenching of the GFP making the autolysosome appear red. The contents, including RFP-LC3B and p62 are then degraded by lysosomal enzymes. b, HEK-293 cells were transfected with CLN3 WT, CLN3Δex7/8 (Δ78), or CLN3Δex5/7/8 (Δ578) expression plasmids. After 44 h, the cells were transfected with the tandem reporter GFP-RFP-LC3 to examine autophagic flux. Cells were treated with DMSO (-) or to induce autophagy, AZD8055 for 20 h then imaged. Shown are representative confocal microcopy images of the formation of autophagosomes (yellow in overlay images) and autolysosomes (red in overlay images) as quantitated in Fig. 1d, n = 10 cells/group. Scale bar, 5 µm.
Extended Data Fig. 3 Lysosome assessment in cells expressing CLN3, CLN3Δex7/8, or CLN3Δex5/7/8.
a, Representative confocal microscopy images of HEK-293 cells expressing CLN3 WT, CLN3Δex7/8, or CLN3Δex5/7/8 and incubated with Lysotracker (red) to visualize lysosomes. Scale bar, 10 µm. b, The number of lysosomes per CLN3 WT, CLN3Δex7/8, or CLN3Δex5/7/8 expressing cells plotted as mean ± s.e.m. n = 20 cells per group. WT CLN3 vs. CLN3Δex7/8, P = 0.797; CLN3Δex7/8 vs. CLN3Δex5/7/8, P = 0.9447; WT CLN3 vs. CLN3Δex5/7/8, P = 0.9447. One way ANOVA, Tukey’s multiple comparisons test; Non-significant (ns).
Extended Data Fig. 4 Normalization of autophagy measurements with CLN3Δex5/7/8 expression.
a, During the initiation of autophagy, LC3I undergoes lipidation and phosphatidylethanolamine (PE) conjugation to form LC3II which is engulfed in a newly formed phagophore with other cargo such as p62, which subsequently forms the autophagosome. Fusion with lysosomes results in formation of the autolysosomes followed by degradation and recycling of resident contents. b, Western blot of protein lysate from HEK-293 cells transfected with CLN3 WT, CLN3Δex7/8 (Δ78), or CLN3Δex5/7/8 (Δ578) expression plasmids in the presence or absence of amino acids. Proteins were separated by SDS-PAGE and probed for LC3B, p62, and phospho-S6K, a marker of autophagy inhibition. α-tubulin was used as a loading control for each sample. Size markers (kilodaltons) are shown at right of gel images. c, Quantitation of p62 abundance relative to α-tubulin in b. Bars are mean ± s.e.m; n = 3 individually treated tissue culture wells per group. WT: fed vs. starvation, P < 0.0001; CLN3Δex7/8: fed vs. starvation, P < 0.0001; CLN3Δex5/7/8: fed vs. starvation, P < 0.0001; WT fed vs. CLN3Δex7/8 fed, P = 0.9999; CLN3Δex7/8 fed vs. CLN3Δex5/7/8 fed, P = 0.6193; WT fed vs. CLN3Δex5/7/8 fed, P = 0.5817; WT starvation vs. CLN3Δex7/8 starvation, P = 0.0497; CLN3Δex7/8 starvation vs. CLN3Δex5/7/8 starvation, P = 0.0324; WT starvation vs. CLN3Δex5/7/8 starvation, P = 0.9941. Two-way ANOVA, Sidak’s multiple comparisons test; *P < 0.05, ****P < 0.0001, or non-significant (ns). d, Immunoblot detection of autophagy related proteins, LC3B, p62, and phospho-S6K in 3 different CLN3+/Δex7/8 and CLN3Δex7/8/Δex7/8 human fibroblast cell lines treated with ASO-C or ASO-28 in the presence of absence of AZD8055-induced autophagy. Size markers (kilodaltons) are shown at right of gel images. e, Quantification of LC3B in d. n = 3 different CLN3Δex7/8 homozygous and n = 3 heterozygous cell lines. +/Δ78 ASO-C: vehicle vs. AZD8055, P = 0.0039; Δ78/Δ78 ASO-C: vehicle vs. AZD8055, P = 0.0017; Δ78/Δ78 ASO-28: vehicle vs. AZD8055, P = 0.0014; +/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-C + vehicle, P = 0.0001; Δ78/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-28 + vehicle, P = 0.0072; +/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-28 + vehicle, P = 0.0811; +/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-C + AZD8055, P = 0.0002; Δ78/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-28 + AZD8055, P = 0.0059; +/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-28 + AZD8055, P = 0.2210. Two-way ANOVA, Sidak’s multiple comparisons test; **P < 0.01, ***P < 0.001. f, Densitometry analysis of p62 normalized to α-tubulin in d. Bars are mean ± s.e.m. n = 3 different CLN3Δex7/8 homozygous and n = 3 heterozygous cell lines. +/Δ78 ASO-C: vehicle vs. AZD8055, P > 0.9999.; Δ78/Δ78 ASO-C: vehicle vs. AZD8055, P = 0.9990; Δ78/Δ78 ASO-28: vehicle vs. AZD8055, P > 0.9999; +/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-C + vehicle, P = 0.9930; Δ78/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-28 + vehicle, P = 0.1074; +/Δ78 ASO-C + vehicle vs. Δ78/Δ78 ASO-28 + vehicle, P = 0.1641; +/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-C + AZD8055, P = 0.9841; Δ78/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-28 + AZD8055, P = 0.0908; +/Δ78 ASO-C + AZD8055 vs. Δ78/Δ78 ASO-28 + AZD8055, P = 0.1600. Two-way ANOVA, Sidak’s multiple comparisons test; p > 0.05 for all comparisons.
Extended Data Fig. 5 Dose-dependent exon 5 skipping with human CLN3-directed ASOs.
a, RT-PCR of RNA isolated from heterozygous CLN3+/Δex7/8 human fibroblast cell line treated with increasing concentrations (3.125 to 200 nM) of ASO-20 or ASO-28, as labeled. The spliced products are labeled at left of gel. Size markers (bp) are shown at right of gel. Quantitation of exon 5 skipping is shown as [Δ578/(Δ578 + Δ78)] x 100. Results reproduce ASO activity observed in homozygous CLN3Δex7/8 human fibroblast cell line shown in Fig. 1i. b, Exon 5 skipped (%) in relationship to the log of the dose is plotted. The half-maximal effective concentration (EC50) was calculated after fitting the data using non-linear regression with a variable slope. c, RT-PCR analysis of exon 5 skipping in three different CLN3 Batten disease patient-derived cell lines homozygous for CLN3Δex7/8 transfected with the indicated ASOs. d, Quantification of the percentage of exon 5 skipped calculated as: [Δ578/(Δ578 + Δ78) x 100]. Bars are mean ± s.e.m. ASO-C (n = 3) vs. ASO-20 (n = 3), P < 0.0001; ASO-C vs. ASO-28 (n = 3), P < 0.0001; ASO-20 vs. ASO-26, P = 0.9216. One-way ANOVA with Tukey’s multiple comparisons test; ****P < 0.0001, non-significant (ns). The spliced products are labeled at left of gel. Size markers (bp) are shown at right of gel.
Extended Data Fig. 6 CLN3Δex7/8 mRNA is a substrate for nonsense-mediated decay.
a, RT-PCR analysis of CLN3Δex7/8 mRNA isolated from a CLN3 Batten disease patient fibroblast cell line transfected with the indicated ASOs and treated with puromycin to inhibit NMD ( + ) or left untreated (-). An SRSF2 mRNA isoform (ENST00000452355.7) that is a known substrate for NMD was analyzed to confirm NMD inhibition with puromycin treatment. β-actin was included as a loading control. These results were reproduced in one additional independent experiment. b, Quantitation of total CLN3 and SRSF2 mRNA in a. Size markers (basepairs) are shown at right of gel images.
Extended Data Fig. 7 ASO dose-dependent exon 5 skipping in mouse cells.
a, Sequence alignment of the most active ASO to the target Cln3 region. Cln3 exonic and intronic nucleotides are displayed as capital and lowercase letters, respectively. b, RT-PCR analysis of exon 5 splicing from RNA extracted from homozygous mCln3Δex7/8 cells transfected with increasing concentrations of ASO-26 (0.391 nM to 200 nM). Spliced products are indicated at left of gel image and size markers (bp) are shown at right of gel. Graph shows results of a single experiment. Untreated (UT) and mock (M) treated controls are included. The graph displays the percent of exon 5 skipped [Δ578/(Δ578 + Δ78) x 100] in relationship to the log of the dose. The half-maximal effective concentration (EC50) was calculated after fitting the data using nonlinear regression, variable slope.
Extended Data Fig. 8 Mouse body weight following ASO-26 treatment.
Body weights of 2 month (a) and 4.5 month (b) old male and female Cln3+/Δex7/8 and Cln3Δex7/8/Δex7/8 mice treated at P1–2 with ASO-C or ASO-26. Bars are mean ± s.e.m; n is shown in graph. 2 month females: +/Δ78 ASO-C vs. Δ78/Δ78 ASO-C, P = 0.6867; Δ78/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.9917; +/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.6420; 2 month males: +/Δ78 ASO-C vs. Δ78/Δ78 ASO-C, P = 0.59999; Δ78/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.8862; +/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.7368; 4.5 month females: +/Δ78 ASO-C vs. Δ78/Δ78 ASO-C, P = 0.0718; Δ78/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.6440; +/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.3740; 4.5 month males: +/Δ78 ASO-C vs. Δ78/Δ78 ASO-C, P = 0.1359; Δ78/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.6663; +/Δ78 ASO-C vs. Δ78/Δ78 ASO-26, P = 0.4463. One-way ANOVA, Tukey’s multiple comparisons test. All comparisons were within sex and age groups; p > 0.05 for all comparisons.
Extended Data Fig. 9 ASO activity in the CNS of ASO-treated mice.
a, RNA extracted from the cortex, thalamus, striatum, brain stem, spinal cord, and kidney of 19-week-old Cln3+/Δex7/8 (+/Δ78) and Cln3Δex7/8/Δex7/8 (Δ78/Δ78) mice treated at P1 or P2 with ASO-C or ASO-26. b, Quantification of exon 5 skipping in (a). Bars are mean ± s.e.m. n = 4 for all but Cln3+/Δex7/8 ASO-C striatum n = 3; *P < 0.05, ****P < 0.0001, one-way ANOVA, Tukey’s multiple comparisons test within each tissue. c, Brain map indicating regions sampled for SCMAS and GFAP immunohistochemistry. The regions numbered 1–5 are the somatosensory cortex, visual cortex, striatum, thalamus, and hippocampus, respectively.
Extended Data Fig. 10 Assessing off-target activity of ASO-26.
a, The predicted ASO-26 binding location in Zmynd8 pre-mRNA is shown. The arrows indicate the primer binding location. b, RT-PCR analysis of RNA extracted from the cortex of Cln3Δex7/8/Δex7/8 mice treated with either ASO-C or ASO-26. The region of the Zmynd8 transcript predicted to be affected by ASO-26 base pairing was amplified by RT-PCR and products were separated on a 6% polyacrylamide gel. Two alternatively spliced Zmynd8 isoforms were detected at equal levels in all samples. RT-PCR of samples were analyzed twice with similar results. c, Quantification of exon 3 skipped shows that ASO-26 treatment does not alter Zmynd8 alternative splicing. Bars are mean ± s.e.m. n = 2 mice.
Supplementary information
Supplementary Information
Supplementary Table 1.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Unprocessed gels.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed gels.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed gels.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed gels.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 7
Unprocessed gels.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 9
Unprocessed gels.
Source Data Extended Data Fig. 10
Statistical source data.
Source Data Extended Data Fig. 10
Unprocessed gels.
Rights and permissions
About this article

Cite this article
Centa, J.L., Jodelka, F.M., Hinrich, A.J. et al. Therapeutic efficacy of antisense oligonucleotides in mouse models of CLN3 Batten disease. Nat Med 26, 1444–1451 (2020). https://doi.org/10.1038/s41591-020-0986-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-0986-1
