
Abstract
Although antiviral drugs and vaccines have reduced the economic and healthcare burdens of influenza, influenza epidemics continue to take a toll. Over the past decade, research on influenza viruses has revealed a potential path to improvement. The clues have come from accumulated discoveries from basic and clinical studies. Now, virus surveillance allows researchers to monitor influenza virus epidemic trends and to accumulate virus sequences in public databases, which leads to better selection of candidate viruses for vaccines and early detection of drug-resistant viruses. Here we provide an overview of current vaccine options and describe efforts directed toward the development of next-generation vaccines. Finally, we propose a plan for the development of an optimal influenza vaccine.
Similar content being viewed by others


Main
One hundred years have passed since the first recorded influenza pandemic was caused by an influenza A(H1N1) virus—the 1918 Spanish flu (Boxes 1 and 2). Since then, there have been three other pandemics caused by A(H2N2), A(H3N2), and A(H1N1)pdm09 viruses—the 1957 Asian flu, the 1968 Hong Kong flu, and the 2009 swine-origin flu, respectively1. Currently, A(H1N1)pdm09 and A(H3N2) viruses together with influenza B virus (Yamagata and Victorian lineages) cause epidemics as seasonal influenza, but A(H1N1) and A(H2N2) viruses have disappeared1.
To understand and contend with influenza virus, a considerable amount of research has been conducted, and this effort has yielded a vast amount of information. For example, functional analysis of influenza virus proteins in vitro has revealed fundamental virological properties of influenza, resulting in the establishment of a method to generate influenza viruses entirely from plasmids2. This method has been and continues to be used to understand the biology of influenza viruses and to improve influenza countermeasures. Many host proteins have now been shown to contribute to virus propagation, revealing part of the complicated virus–host interaction3,4. Viral proteins and amino acid residues involved in the pathogenicity of influenza virus have also been identified, and the experimental procedures used to assess them are now well-established, leading to rapid risk assessment of newly emerged influenza viruses5,6,7. Several neuraminidase (NA) and polymerase inhibitors, which target virus proteins, have been developed and are efficacious when used early after onset, and rapid influenza diagnostic kits, which can provide results in 5–20 minutes, are now also available8,9,10,11. Seasonal influenza vaccines are available prior to every influenza season, and prepandemic vaccines against particular virus subtypes with pandemic potential have also been prepared12. Nevertheless, the control of seasonal influenza remains suboptimal, and there is always the risk of a pandemic caused by a virus to which the majority of human populations have no immunity.
To understand virus properties, viral genomic sequences have been analyzed since the late 1970s using the Sanger sequencing method, also known as the dideoxy chain termination method13. Recently, deep-sequencing technology has allowed us to determine the whole genomic sequence of many isolates, resulting in the accumulation of a large number of virus sequences in public databases. This wealth of information now makes it possible to monitor viruses circulating worldwide14,15, to predict virus fitness in silico16, and to assess the pandemic risk of virus isolates17.
Here, we review the understanding of viral evolution and spread and the current vaccine situation, and we describe future prospects for the development of next-generation vaccines.
Influenza virus and current epidemics
Influenza viruses cause a respiratory illness with symptoms such as fever, cough, sore throat, runny nose, muscle or body aches, headaches, and/or gastrointestinal symptoms (vomiting and diarrhea). The virus annually causes 3‒5 million severe cases, 0.3‒0.6 million deaths, and subsequent economic losses18. Currently, the influenza A virus subtypes H1N1pdm09 and H3N2, as well as influenza B viruses of the Yamagata and Victoria lineages, are as globally prevalent among humans as seasonal influenza viruses (Box 1). Global year-round surveillance is conducted by the World Health Organization (WHO) Global Influenza Surveillance and Response System (GISRS), which includes the National Influenza Centres, the WHO Collaborating Centres for Reference and Research on Influenza, and the Essential Regulatory Laboratories, to monitor changes in the virus genome, especially in hemagglutinin (HA) and NA (http://www.who.int/influenza/gisrs_laboratory/flunet/en/). On the basis of the phylogenetic similarity of the nucleotide sequence of HA, epidemic viruses are classified into a ‘clade’ or ‘subclade.’ A real-time snapshot of the current populations of these viruses is available at the website nextstrain.org15.
Emergence and subsequent evolution of pandemic viruses in humans
Pandemic influenza is caused by the emergence of a virus with an HA protein to which the majority of human populations do not have immunity19. Previously, it was thought that a pandemic occurs when a virus whose HA subtype is different from that of viruses circulating in humans emerges; however, this concept was challenged when the A(H1N1)pdm09 virus caused a pandemic in 2009 even though A(H1N1) and A(H3N2) viruses were cocirculating. Influenza A viruses of a variety of subtypes are naturally maintained in avian species, especially aquatic birds, and are the typical source of the current HA. In contrast, influenza B viruses are unlikely to cause a pandemic because their antigenic diversity is limited. Reassortment (i.e., the exchange of genes between two or more influenza viruses upon co-infection of cells) between human, swine, and/or avian viruses in pigs and the direct interspecies transmission of an avian virus to humans led to the latest four pandemics1 (Box 2). One of the most important facts that we have learned from these past pandemics is that no one knows when, where, or which subtype of influenza virus will cause the next pandemic.
After each pandemic, all four pandemic viruses continued to circulate in humans as a seasonal influenza virus after competing out previous seasonal viruses. Since inhibitory antibodies against HA and NA are usually elicited upon influenza virus infection in infected individuals20,21,22, it is viruses with amino acid mutations in HA and NA that are responsible for antigenic changes for evasion from such antibodies, so-called ‘antigenic drift' (Fig. 1). The mutations in HA mostly accumulate around the receptor-binding site (RBS) because antibodies that recognize the area around the RBS efficiently inhibit the binding of HA to its receptor, resulting in the neutralization of virus infectivity23,24. The five major antigenic sites, Ca1, Ca2, Cb, Sa, and Sb for H1 HA and A through E for H3 HA, have been mapped by X-ray crystallography, comparative sequence analysis, and characterization of mutant viruses that escaped from neutralizing mouse monoclonal antibodies25,26,27,28. Antigenic cartography suggests that antigenic drift of human influenza viruses occurs in clusters; while nucleotide changes continue to occur, clusters of antigenically similar variants exist for several years until they are replaced by viruses that form a novel cluster29,30, meaning that the genetic evolution is continuous, whereas antigenic evolution is punctuated. The ‘cluster-transitions’ of A(H3N2) virus over a 35-year period were predominantly caused by single amino acid substitutions that occurred at only seven positions (position 145 at antigenic site A and positions 155, 156, 158, 159, 189, and 193 at antigenic site B) adjacent to the RBS31. Similarly, mutations in NA are frequently identified around the enzymatic active center32,33. Antibodies that recognize these epitopes interfere with the sialidase activity of NA, resulting in the suppression of virus release from infected cells34. Studies with monoclonal antibodies and amino acid sequence analysis have revealed two to three antigenic sites in the NA protein35.

Debbie Maizels/Springer Nature
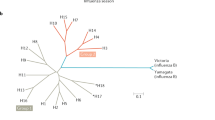
Immunologically naive human populations are infected with an influenza virus (i). Infected individuals acquire immunity against influenza virus (yellow) (ii). Viruses accumulate amino acid mutations in their antigenic HA during replication (iii). Some individuals who do not possess immunity against the initial virus are infected by the mutated virus (iv). Infected individuals acquire immunity against this virus (purple) (v). The virus further accumulates amino acid mutations in its HA (vi). The remaining naive individuals are attacked by this further mutated virus (vii). The majority of the individuals in human populations eventually acquire immunity against these viruses (yellow, purple, orange). They are then protected from influenza viruses with similar antigenicity (viii). The virus obtains one or two mutations in its HA that substantially alter antigenicity (i.e., antigenic drift) (xi). The antigenically drifted virus can infect individuals who possessed immunity against the previously circulating viruses (x). The individuals infected with the antigenically drifted virus mount immunity to this virus (blue). The cycle continues (xi). b, Real-world tracking of influencza viral epidemics allows the development of appropriate vaccines. Note that the virus evoles gradually, whereas antigenic shift occurs in steps. This is a screenshot of a phylogenic tree based on the HA sequences of human A(H3N2) viruses isolated between 2006 and 2018 from next strain (https://nextstrain.org/). Subclades (3b, 3c, 3c2, 3c3, and 3c2A, etc.) are indicated by grey letters, and antigenic advance of isolates is indicated by each color. Vaccine seed A(H3N2) viruses indicated in the tree were changed in response to antigenic advance of circulating virusese. The labels are the names of virus isolates. For example, in A/Texas/50/2012, ‘A’ means influenza A virus, ‘50’ is sample number, and ‘2012’ is collection year.
Current influenza vaccines
To reduce the burden attributed to seasonal and pandemic influenza, multiple approaches, including vaccines and antiviral drugs, have been developed. Since a fully effective vaccine, if available, would be able to prevent influenza completely, vaccination is an appropriate option to combat influenza virus. Currently, three kinds of vaccines (inactivated, live attenuated, and recombinant HA vaccines) are licensed in various countries, and each type of vaccine has advantages and drawbacks36. For all of these vaccines, the vaccine seed viruses must be replaced periodically to match their antigenicity to that of the circulating viruses. Since antigenic mismatch causes low vaccine efficacy, the WHO biannual influenza vaccine composition meetings (one for the Northern hemisphere and the other for the Southern hemisphere) try to select the correct ones on the basis of the genetic and antigenic characteristics of the circulating viruses and epidemiologic information from individual countries. Since the vaccine seed viruses are determined more than 6 months before each epidemic season (http://www.who.int/influenza/vaccines/virus/recommendations/consultation201802/en/), antigenic mismatch between vaccine candidates and circulating strains occurs occasionally37.
Inactivated vaccines
Inactivated vaccine, produced by growing the vaccine seed virus in chicken embryonated eggs, is the most popular approach in the world because of relatively low production costs and high safety. Vaccinations with inactivated vaccines begin at 6‒12 months of age, and annual vaccinations are needed because the immunity conferred by the vaccine does not last long38. There are three types of inactivated vaccines: whole-virion vaccines, split-virion vaccines, and subunit vaccines. Whole-virion vaccine is prepared by purification of virions that have been chemically inactivated with formaldehyde or β-propiolactone. In the split-virion vaccine, the virus envelope of the whole virion is disrupted by diethyl ether or detergent treatment. Subunit vaccines contain HA and NA that are further purified by exclusion of the viral ribonucleoprotein (vRNP), M1, and viral envelope (lipid). Despite low immunogenicity and a narrow range of protection, split-virion and subunit vaccines are used more commonly than whole-virion vaccines to vaccinate humans against seasonal influenza. Another key issue for inactivated vaccines is that influenza A virus, especially the recent A(H3N2) virus, requires many passages in eggs to achieve high titers because the initial isolates replicate poorly in eggs. Excessive passages in eggs can change the antigenicity of HA, resulting in an antigenic mismatch with the epidemic isolates39,40,41.
To avoid egg-adaptive mutations in HA, cultured cell lines (such as Madin-Darby canine kidney (MDCK) and Vero cells) can be used for virus propagation42. However, the titers of vaccine seed viruses in such cell lines grown under serum- and animal-component-free conditions and in suspension or a bioreactor are lower than those in eggs, resulting in high cost and low productivity43. Therefore, the use of cell-culture-based inactivated seasonal vaccines has been limited.
Live attenuated vaccines
Live attenuated vaccines are available in the United States, Canada, and several European countries. Vaccines derived from cold-adapted and temperature-sensitive master donor viruses44,45,46 are propagated in eggs, causing egg-adaptive mutations in HA. Because live attenuated vaccines mimic a natural infection without causing major adverse reactions, they can elicit both IgA, which is the principal isotype in secretions at the mucous membrane and which operates mainly on epithelial surfaces, in the upper respiratory tract as well as IgG, which is the principal isotype in blood and extracellular fluid and which operates mainly in body tissues, in serum, providing cross-reactive immune responses at the initial replication site47,48. However, the live attenuated vaccine is not recommended for use in children younger than 2 years of age, pregnant women, and people with certain underlying illnesses or a compromised immune system because the vaccine viruses may replicate to higher titers in these individuals, leading to some side effects.
Recombinant HA vaccines
The recombinant HA vaccine is produced by a recombinant-protein-expressing system using insect cells and baculovirus49 and is approved by the Food and Drug Administration (FDA) for use in the United States. Since this system does not use live influenza viruses, HA protein is obtained that lacks the unwanted mutations that can be introduced during egg adaptation. Furthermore, recombinant HA vaccine can be manufactured within 2 months, indicating that this vaccine would be suitable for the prevention of pandemic influenza viruses. Although the mechanism of action of this vaccine is similar to that of inactivated vaccines, the commercial formulation of the recombinant HA vaccine contains three times the amount of HA as the inactivated influenza vaccines to induce antibody titers equivalent to those obtained with conventional inactivated vaccines50. Since the elicited immunity is HA- and strain-specific, the HA must be updated frequently to match the antigenicity of the epidemic strains. The recombinant HA vaccine is limited to use in individuals 18‒49 years of age because of its low immunogenicity, especially in children.
Development of next-generation vaccines
Although vaccines are used in many countries, seasonal influenza epidemics have not been controlled. To improve the effectiveness of vaccines, advances must be made in five major areas: selection of the vaccine seed virus, targeting the vaccine, use of cultured cells instead of eggs for vaccine virus preparation, increasing the NA content of vaccines, and development of novel classes of adjuvants.
Selection of vaccine seed virus
To improve vaccine selection, in silico and in vitro studies have been conducted. Current epidemics can be visualized by integrating sequence data with epidemiologic information14 or by the continuous updating of databases to monitor the rise and decline of virus clades15,51 (Fig. 2). In silico modelling using past epidemic patterns together with information on viral fitness16 or the relative distances of amino acid sequences in the multidimensional scaling-constructed 3D space52 are used to predict the future direction of influenza virus evolution. To model which viruses may circulate in the future, viruses possessing random mutations in the HA head are first generated by reverse genetics. Of these, the viruses that could escape from neutralizing antibodies against HA are identified by using antisera obtained from ferrets that are experimentally infected with the parental virus, or sera obtained from humans who were exposed to influenza virus infection or vaccination53. These approaches allow the antigenicity of future epidemic strains to be determined and, in some cases, the exact amino acid changes that may occur can be predicted. Therefore, scientists are getting closer to identifying viruses that are antigenically similar to those that may circulate in nature before they emerge.

Debbie Maizels/Springer Nature
Real-time tracking of influenza A is now possible, which allows tracking of strains across the globe. This is a screen shot of a phylogenic tree based on the HA sequences of human A(H3N2) viruses isolated between 2016 and 2018 generated at https://nextstrain.org/. Subclades (3c2, A1, A1b, and A1a, etc.) are indicated by grey letters, and different colors indicate where the isolate was isolated.
The next challenge for the selection of better vaccine components is to identify emerging viruses with antigenic drift. Antigenically similar viruses circulate in humans for several years without antigenic change31. During this period, the viruses first accumulate amino acid mutations in their HA that do not affect antigenicity and then acquire mutations that do affect the antigenicity of the virus, resulting in the emergence of antigenically drifted viruses (Fig. 1). Therefore, we need to learn what triggers the emergence of the latter amino acid changes. Studies on the antibody landscape (i.e., population dynamics of the levels of antibodies against the circulating strains) may help to solve this problem.
Vaccine targeting
The Center for Disease Control (CDC) recommends influenza vaccinations for all age groups (https://www.cdc.gov/flu/protect/whoshouldvax.htm). Individuals with a relatively higher risk for influenza, such as young children, the elderly, pregnant women, and people with chronic medical conditions, are highly encouraged to get vaccinated54,55,56. For these high-risk groups, vaccination tailored to specific targets may improve protection. For young children, the inactivated vaccine and live attenuated vaccine are available for individuals >6 months of age and >2 years of age, respectively (see above). Although annual vaccination for those who are 6 months of age and older is recommended to reduce the risk of influenza, it is unclear how best to induce ‘good’ immunologic imprinting in these individuals (see below). For pregnant women, vaccination benefits both the pregnant woman and her unborn baby54.
Improvement of cell-based vaccine productivity
To address problems with low productivity, efforts have been made in two distinct areas: modification of the virus and amelioration of the cells in which it is produced. For virus modification, several sets of a vaccine backbone are prepared by optimizing the polymerase activity and efficiency of genome packaging and virion release of the influenza A or B vaccine viruses to achieve a high virus titer in MDCK and/or Vero cells57,58,59,60. A mutation that increases the fidelity of the virus polymerase may be useful for genetic stability of the virus for vaccine production61,62. Such backbones, together with HA and NA segments derived from circulating isolates, can be utilized for virus candidate production by using plasmid-driven reverse genetics2. For cell amelioration, researchers pick up high-virus-producing clones from parental cells63, downregulate host protein expression that suppresses virus growth64, and upregulate expression of human-type virus receptor65,66. Although these improvements show promise, they have not yet been incorporated into actual vaccine production.
NA content
The protection afforded by current inactivated vaccines is thought to be primarily mediated by HA because HA is a major target for protective antibodies. Therefore, the HA content of inactivated vaccines is standardized and measured. Despite the fact that antibody responses to NA have been shown to be the only independent immune correlate of all assessed measures of protection in human challenge models67, the NA content of inactivated vaccines is not quantified and is suboptimal, resulting in a lack of immune response against NA in vaccinees68. In infected patients, NA can elicit protective antibodies, most of which possess neuraminidase inhibition (NI) activity68; some antibodies without NI activity also protect against influenza infection via Fc-mediated effector cell activation69. Although antigenic drift and immunologic imprinting of NA have been reported70,71, only NA antibodies that recognize the epitopes around the enzymatic active site and inhibit sialidase activity have been studied. Therefore, there is a need for analyses of antibodies against the NA head that lack sialidase inhibitory activity and of anti-NA stalk antibodies to fully understand the importance of NA as a vaccine antigen. In addition, a cross-reactive anti-NA antibody that binds and inhibits N1 through N9 NA activity was shown to be partially protective against H1N1 and H3N2 virus infection in mice72, suggesting that an NA-targeted vaccine may have the potential to induce cross-protective antibodies and that the NA content in inactivated vaccines should be increased and standardized.
Adjuvants
Another improvement in vaccine efficacy may come from novel classes of adjuvant. An adjuvant is a substance that is formulated as part of a vaccine to enhance its ability to induce protection via activation of the immune system, allowing the antigens in vaccines to induce long-term protective immunity. The current adjuvanted vaccines normally cause local and general side effects, including pain, fatigue, headache, and myalgia, more frequently than nonadjuvanted vaccines because the adjuvant fundamentally promotes immune responses by mimicking the infection and causing inflammation. Mild adverse effects are acceptable, but severe adverse effects should be avoided. The severe adverse reaction of an increased risk of narcolepsy has been reported to be associated with the currently licensed adjuvant AS03 (refs. 73,74).
To reduce the possibility of the occurrence of serious adverse effects, researchers have developed novel classes of adjuvant with a clear mechanism of action. Toll-like receptor (TLR) ligands are the best understood of these adjuvants. TLRs are members of a family of pattern recognition receptors that recognize common motifs of pathogens. TLR4 agonists MPLA (monophosphoryl lipid A) and GLA (glucopyranosyl lipid adjuvant), a TLR7/8 agonist (imiquimod), a TLR3 agonist (rintatolimod), a TLR9 agonist (CpG ODN (CpG oligodeoxynucleotide)), and a TLR5 agonist (flagellin) have been evaluated as influenza vaccine adjuvants75. Cytokines are well-characterized cell signaling molecules. Since cytokine induction is an essential action for most adjuvants, representative cytokines involved in immune responses, such as the T cell activator IL-2, the dendritic cell activator granulocyte-macrophage colony-stimulating factor (GM-CSF), and type I interferon, have been incorporated as adjuvants into vaccines that are currently being developed12.
Other formulations and immunostimulators have been developed as adjuvants, but their mechanisms of action have not yet been characterized; the simplest approach to improving the host immune response via an adjuvant could be usage of the whole virion as the vaccine antigen. Whole-virion vaccines, but not split vaccines, elicit high neutralizing antibodies in the early T-cell-independent response, which requires B-cell–intrinsic TLR7 signaling activated by viral RNAs within the whole-virion vaccine76.
Evaluation of host immune responses to vaccines
Vaccine efficacy is assessed according to the level of protection from infection the vaccine provides. However, surrogates, such as hemagglutinin-inhibition (HI) antibody titers in sera and virus-specific cytotoxic T lymphocyte (CTL) levels have been used to evaluate vaccine immunogenicity. Since a serum HI antibody titer of ≥ 1:40 is associated with a significant reduction in influenza incidence, serum HI antibody titer induction is used as a measure of vaccine efficacy in clinical trials77. The inactivated vaccine elicits HI antibodies against viruses that vaccinees have encountered over their lifetime, not just the antigens in the vaccines, an effect known as the ‘back-boost’78. Such serologic evaluation can measure the quantity of HI antibodies, especially the most potent neutralizing antibodies that recognize the epitopes around the HA RBS.
Recent studies have revealed that infection- and vaccine-induced human in vitro neutralizing and non-neutralizing antibodies against the HA stem, which mostly recognize heterosubtypic HA79,80,81, show in vivo protective efficacy via Fcγ-receptor-medicated activation of natural killer (NK) cells (antibody-dependent cellular cytotoxicity; ADCC), macrophages (antibody-dependent cellular phagocytosis; ADCP), and neutrophils (antibody-dependent neutrophil-mediated phagocytosis; ADNP)69,82,83. Antibodies against the HA head or stem also inhibit virus particle release from infected cells81,84. Thus, antibodies other than HI antibodies should be measured to evaluate vaccine immunogenicity in future trials. Accordingly, it is important to develop methods to evaluate different types of immunity that can serve as immune correlates for protection.
Since vaccination induces antibodies that protect vaccinees by different mechanisms, analyses using sera that contain a mixture of many different antibodies are not useful for developing a mechanistic understanding of vaccine protection. Antibody responses have been qualitatively analyzed by determining B cell receptor sequences to reveal the antibody repertoire at the molecular level. An unbiased antibody repertoire analysis revealed that the inactivated vaccine elicits antibody production from memory and naive B cells85. Of the elicited antibodies, many cross-reactive neutralizing clones that recognized the HA RBS and many cross-reactive non-neutralizing protective clones that recognized the lateral surface of the HA head were detected in some individuals85,86. However, the antibody response to a vaccine differs among individuals owing to differences in their history of influenza infection and vaccination87,88,89.
Generation of broadly protective vaccines
Current inactivated vaccines provide some protection to vaccine recipients from viruses that are antigenically similar to the vaccine viruses. However, such vaccines fail to suppress infections caused by antigenically drifted viruses and offer no protection against an antigenic shifted virus that has the potential to cause a pandemic. Therefore, there is a need for a vaccine capable of inducing immune responses that last for a long time and that protect against a wide range of viruses, ideally all influenza A and B viruses90. Several approaches have been taken to produce such vaccines (so-called universal vaccines) based on the concept of inducing immune responses against the conserved protective epitopes in virus proteins. The targets of universal vaccine candidates include the HA stem, the RBS of HA, the extracellular domain of M2 (M2e), and the CTL epitopes in M1 and NP36.
Although the antigenicity of the HA head varies between HA subtypes, that of the HA stem is highly conserved among HA group members79,91. Several epitopes in the HA stem are common across groups 1 and 2 (ref. 80), and an epitope conserved in both type A and B viruses has also been reported81. Antibodies against the stem are typically heteroreactive and suppress virus replication by inhibiting membrane fusion and virus release as well as the activation of Fc-region-mediated cytotoxicity84,92,93. Therefore, several approaches have been proposed to elicit anti-HA stem antibodies, including immunization with headless HA94,95,96,97, sequential chimeric or heterosubtypic HA98,99,100, synthetic fragments or peptides of the HA stem101,102, and hyperglycosylated HA103.
M2 is a relatively conserved tetrameric type III transmembrane protein that functions as a proton-selective ion channel. M2e has been extensively investigated as a target for a universal vaccine. Antibodies against M2e do not interfere with virus entry but prevent virus release and activate effector cells via an Fc–receptor interaction104. Since M2e per se is poorly immunogenic, various strategies such as multimerization, display on virus-like particles or phages, and fusion with a carrier protein or a protein with adjuvant activity are being tested to improve the host immune response105. Animals vaccinated with M2e were shown to be protected from homologous and heterologous challenge infection105,106. Although several kinds of M2e vaccine have been evaluated in early-phase clinical trials, no M2e vaccine is as of yet available on the market. To overcome some of the limitations of the M2e-based vaccines, combination with other conserved proteins is now under consideration105.
Despite the high variability of the HA head, the RBS is functionally conserved because sialic acid receptor recognition is an essential step for influenza virus entry. Antibodies against the RBS mimic the binding mode of sialic acid to some extent, resulting in a high cross-neutralizing capability107,108,109. Although the RBS could be a target for a broadly protective vaccine, such a vaccine is not being actively investigated because of the lack of an optimally designed antigen. However, efforts towards this goal are underway, as are efforts towards the development of antiviral drugs targeting the RBS110,111.
NP and M1 are highly conserved among influenza A viruses; however, they are generally considered unsuitable targets for antibody-inducing vaccines owing to their lack of exposure on the virion surface, although NP has been detected on the cell surface112. Therefore, the conserved epitopes in these proteins are targets for CTLs, resulting in a broadly cross-reactive response. In fact, NP is the major target for the CTL response in humans113. Since activated CD8+ T cells attack infected cells and enhance virus clearance, the CTL-inducing vaccines reduce disease severity and mortality but do not prevent infection. A modified vaccinia Ankara (MVA) expressing NP and M1 (MVA-NP + M1)114 or a mixture of synthetic polypeptides derived from M1 and NP115 has been used to induce CTL activation. These vaccines were evaluated in phase 1b or 2a trials and induced a good CTL response in humans116,117.
If the next-generation vaccines are expected to induce antibodies possessing HI activity, it would seem to be appropriate to evaluate these vaccines by using the current HI assay. However, most of the next-generation vaccines currently under development target areas other than the major antigenic sites of the HA head. Therefore, HI antibody titers are not a suitable measure for assessing such vaccines. Although antibody titers against the HA stem, M2e, or NA, or CTL activation against NP or M1 can be measured in humans and animals, we do not know whether they can serve as immune correlates of protection. Moreover, the evaluation method and threshold values used differ among vaccines. Therefore, regulatory science to control and evaluate next-generation vaccines needs to be established.
Optimal protection by understanding imprinting
Many candidate next-generation vaccines are under investigation in clinical or preclinical trials. One essential issue, ‘immunologic imprinting’ (or the so-called original antigenic sin), remains to be understood and managed. In 1960, it was proposed that the initial exposure to an influenza virus affects the antibody response to subsequent virus exposures118.
Recent epidemiological research regarding H5N1 and H7N9 viruses has demonstrated that lifelong immunologic imprinting, which is elicited by infection with the influenza strain circulating during one’s childhood, helps protect against unfamiliar HA subtypes from the same HA group119. This immunologic imprinting varies among individuals depending on the year of their birth and the virus strains they encountered, and it likely impacts how individuals will respond to the antigens of next-generation vaccines as well as current inactivated split vaccines (Fig. 3). Therefore, this necessitates a complete understanding of immunological imprinting by analyzing its establishment in an individual’s childhood. It may be possible to optimally immunologically imprint individuals in childhood and to induce optimal immune responses in adults and the elderly by avoiding the original antigenic sin. Moreover, scientists must establish animal models for evaluation of immunologic imprinting; most animal experiments are conducted using animals that have never been infected with influenza virus. Optimal immunologic imprinting by vaccination of animals needs to be established to avoid unwanted immunologic imprinting.

Debbie Maizels/Springer Nature
Immunologic imprinting varies from generation to generation because the circulating influenza A viruses in childhood differ. The first encounters of individuals born between 1918 and 1957, between 1957 and 1968, between 1968 and 1977, between 1977 and 2009, or after 2009 with an influenza virus would have been with H1N1, H2N2, H3N2, H1N1 or H3N2, or H1N1pdm09 or H3N2 virus, respectively. Even though the virus subtypes were identical between these periods, the antigenicity of the viruses changed over time. Therefore, infections during childhood immunologically imprinted the infected individuals in a variety of ways. This imprinting affects immune responses to subsequent virus infection and vaccination.
Concluding remarks
Many kinds of next-generation vaccines are under development for clinical use. To establish truly universal vaccines, several of these vaccines may need to be combined. Although it will not be easy to develop a universal influenza vaccine, it may be possible with time, money, wisdom, and collaboration between laboratories, companies, and countries.
References
Neumann, G., Noda, T. & Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459, 931–939 (2009).
Neumann, G. et al. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl Acad. Sci USA 96, 9345–9350 (1999).
Watanabe, T. et al. Influenza virus–host interactome screen as a platform for antiviral drug development. Cell Host Microbe 16, 795–805 (2014).
Tripathi, S. et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Cauldwell, A. V., Long, J. S., Moncorge, O. & Barclay, W. S. Viral determinants of influenza A virus host range. J. Gen. Virol. 95, 1193–1210 (2014).
Yamayoshi, S. et al. Enhanced replication of highly pathogenic influenza A(H7N9) virus in humans. Emerg. Infect. Dis. 24, 746–750 (2018).
Imai, M. et al. A highly pathogenic avian H7N9 influenza virus isolated from a human is lethal in some ferrets infected via respiratory droplets. Cell Host Microbe 22, 615–626 e618 (2017).
Sakai-Tagawa, Y. et al. Reactivity and sensitivity of commercially available influenza rapid diagnostic tests in Japan. Sci. Rep. 7, 14483 (2017).
Furuta, Y. et al. Favipiravir (T–705), a novel viral RNA polymerase inhibitor. Antiviral Res. 100, 446–454 (2013).
Omoto, S. et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 8, 9633 (2018).
Kim, C. U., Chen, X. & Mendel, D. B. Neuraminidase inhibitors as anti-influenza virus agents. Antivir. Chem. Chemother. 10, 141–154 (1999).
Tregoning, J. S., Russell, R. F. & Kinnear, E. Adjuvanted influenza vaccines. Hum. Vaccin. Immunother. 14, 550–564 (2018).
Both, G. W. & Air, G. M. Nucleotide sequence coding for the N-terminal region of the matrix protein influenza virus. Eur. J. Biochem. 96, 363–372 (1979).
Nolte, N., Kurzawa, N., Eils, R. & Herrmann, C. MapMyFlu: visualizing spatio-temporal relationships between related influenza sequences. Nucleic Acids Res. 43, W547–551 (2015).
Hadfield, J., et al. Nextstrain: real-time tracking of pathogen evolution. Preprint at https://doi.org/10.1101/224048 (2017).
Luksza, M. & Lassig, M. A predictive fitness model for influenza. Nature 507, 57–61 (2014).
Russell, C. A. et al. Improving pandemic influenza risk assessment. eLife 3, e03883 (2014).
Iuliano, A. D. et al. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391, 1285–1300 (2018).
Itoh, Y. et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460, 1021–1025 (2009).
Potter, C. W. & Oxford, J. S. Determinants of immunity to influenza infection in man. Br. Med. Bull. 35, 69–75 (1979).
Hobson, D., Curry, R. L., Beare, A. S. & Ward-Gardner, A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J. Hyg. (Lond). 70, 767–777 (1972).
Wrammert, J. et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208, 181–193 (2011).
Gerhard, W., Yewdell, J., Frankel, M. E. & Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290, 713–717 (1981).
Atassi, M. Z. & Webster, R. G. Localization, synthesis, and activity of an antigenic site on influenza virus hemagglutinin. Proc. Natl Acad. Sci. USA 80, 840–844 (1983).
Webster, R. G. & Laver, W. G. Determination of the number of nonoverlapping antigenic areas on Hong Kong (H3N2) influenza virus hemagglutinin with monoclonal antibodies and the selection of variants with potential epidemiological significance. Virology 104, 139–148 (1980).
Wiley, D. C., Wilson, I. A. & Skehel, J. J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Caton, A. J., Brownlee, G. G., Yewdell, J. W. & Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31, 417–427 (1982).
Xu, R. et al. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328, 357–360 (2010).
Smith, D. J. et al. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376 (2004).
Bedford, T. et al. Integrating influenza antigenic dynamics with molecular evolution. eLife 3, e01914 (2014).
Koel, B. F. et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science 342, 976–979 (2013).
Air, G. M., Els, M. C., Brown, L. E., Laver, W. G. & Webster, R. G. Location of antigenic sites on the three-dimensional structure of the influenza N2 virus neuraminidase. Virology 145, 237–248 (1985).
Gulati, U. et al. Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98). J. Virol. 76, 12274–12280 (2002).
Colman, P. M. & Ward, C. W. Structure and diversity of influenza virus neuraminidase. Curr. Top. Microbiol. Immunol. 114, 177–255 (1985).
Air, G. M. & Laver, W. G. The neuraminidase of influenza virus. Proteins 6, 341–356 (1989).
Rajao, D. S. & Perez, D. R. Universal vaccines and vaccine platforms to protect against influenza viruses in humans and agriculture. Front. Microbiol. 9, 123 (2018).
Xie, H. et al. H3N2 mismatch of 2014–15 northern hemisphere influenza vaccines and head-to-head comparison between human and ferret antisera derived antigenic maps. Sci. Rep. 5, 15279 (2015).
Young, B., Sadarangani, S., Jiang, L., Wilder-Smith, A. & Chen, M. I. Duration of influenza vaccine effectiveness: a systematic review, meta-analysis, and meta-regression of test-negative design case-control studies. J. Infect. Dis. 217, 731–741 (2018).
Zost, S. J. et al. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl Acad. Sci. USA 114, 12578–12583 (2017).
Wu, N. C. et al. A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine. PLoS Pathog. 13, e1006682 (2017).
Raymond, D. D. et al. Influenza immunization elicits antibodies specific for an egg-adapted vaccine strain. Nat. Med. 22, 1465–1469 (2016).
Feng, S. Z., Jiao, P. R., Qi, W. B., Fan, H. Y. & Liao, M. Development and strategies of cell-culture technology for influenza vaccine. Appl. Microbiol. Biotechnol. 89, 893–902 (2011).
Hegde, N. R. Cell culture-based influenza vaccines: a necessary and indispensable investment for the future. Hum. Vaccin. Immunother. 11, 1223–1234 (2015).
Ghendon, Y. Z. et al. Recombinant cold-adapted attenuated influenza A vaccines for use in children: molecular genetic analysis of the cold-adapted donor and recombinants. Infect. Immun. 44, 730–733 (1984).
Jin, H. et al. Multiple amino acid residues confer temperature sensitivity to human influenza virus vaccine strains (FluMist) derived from cold-adapted A/Ann Arbor/6/60. Virology 306, 18–24 (2003).
Hoffmann, E. et al. Multiple gene segments control the temperature sensitivity and attenuation phenotypes of ca B/Ann Arbor/1/66. J. Virol. 79, 11014–11021 (2005).
Suzuki, T. et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc. Natl Acad. Sci. USA 112, 7809–7814 (2015).
Hoft, D.F. et al. Comparisons of the humoral and cellular immune responses induced by live attenuated influenza vaccine and inactivated influenza vaccine in adults. Clin. Vaccine. Immunol . 24, e00414–e00416 (2017).
Cox, M. M. & Hollister, J. R. FluBlok, a next generation influenza vaccine manufactured in insect cells. Biologicals 37, 182–189 (2009).
Cox, M. M., Patriarca, P. A. & Treanor, J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir. Viruses 2, 211–219 (2008).
Neher, R. A. & Bedford, T. nextflu: real-time tracking of seasonal influenza virus evolution in humans. Bioinformatics 31, 3546–3548 (2015).
Ito, K. et al. Gnarled-trunk evolutionary model of influenza A virus hemagglutinin. PLoS One 6, e25953 (2011).
Li, C. et al. Selection of antigenically advanced variants of seasonal influenza viruses. Nat. Microbiol. 1, 16058 (2016).
Sakala, I. G., Honda-Okubo, Y., Fung, J. & Petrovsky, N. Influenza immunization during pregnancy: benefits for mother and infant. Hum. Vaccin. Immunother. 12, 3065–3071 (2016).
Schaffner, W., Chen, W. H., Hopkins, R. H. & Neuzil, K. Effective immunization of older adults against seasonal influenza. Am. J. Med. 131, 865–873 (2018).
Rizzo, C., Rezza, G. & Ricciardi, W. Strategies in recommending influenza vaccination in Europe and US. Hum. Vaccin. Immunother. 14, 693–698 (2018).
Ping, J. et al. Development of high-yield influenza A virus vaccine viruses. Nat. Commun. 6, 8148 (2015).
Ping, J., Lopes, T. J., Neumann, G. & Kawaoka, Y. Development of high-yield influenza B virus vaccine viruses. Proc. Natl Acad. Sci. USA 113, E8296–E8305 (2016).
Dormitzer, P. R. et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 5, 185ra168 (2013).
Hu, W. et al. A Vero-cell-adapted vaccine donor strain of influenza A virus generated by serial passages. Vaccine 33, 374–381 (2015).
Cheung, P. P. et al. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat. Commun. 5, 4794 (2014).
Naito, T., et al. Generation of a genetically stable high-fidelity influenza vaccine strain. J. Virol. 91, e01073-16 (2017).
Liu, J., Mani, S., Schwartz, R., Richman, L. & Tabor, D. E. Cloning and assessment of tumorigenicity and oncogenicity of a Madin-Darby canine kidney (MDCK) cell line for influenza vaccine production. Vaccine 28, 1285–1293 (2010).
Hamamoto, I., Takaku, H., Tashiro, M. & Yamamoto, N. High yield production of influenza virus in Madin Darby canine kidney (MDCK) cells with stable knockdown of IRF7. PLoS One 8, e59892 (2013).
Hatakeyama, S. et al. Enhanced expression of an alpha2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J. Clin. Microbiol. 43, 4139–4146 (2005).
Matrosovich, M., Matrosovich, T., Carr, J., Roberts, N. A. & Klenk, H. D. Overexpression of the alpha-2,6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J. Virol. 77, 8418–8425 (2003).
Park, J. K., et al.Evaluation of preexisting anti-hemagglutinin stalk antibody as a correlate of protection in a healthy volunteer challenge with influenza A/H1N1pdm virus. MBio 9, e02284-17 (2018).
Chen, Y. Q. et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429 e410 (2018).
DiLillo, D. J., Palese, P., Wilson, P. C. & Ravetch, J. V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 126, 605–610 (2016).
Sandbulte, M. R. et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl Acad. Sci. USA 108, 20748–20753 (2011).
Rajendran, M. et al. Analysis of anti-influenza virus neuraminidase antibodies in children, adults, and the elderly by elisa and enzyme inhibition: evidence for original antigenic sin. MBio 8, e02281-16 (2017).
Doyle, T. M. et al. Universal anti-neuraminidase antibody inhibiting all influenza A subtypes. Antiviral Res. 100, 567–574 (2013).
Nohynek, H. et al. AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS One 7, e33536 (2012).
Miller, E. et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. BMJ 346, f794 (2013).
Weinberger, B. Adjuvant strategies to improve vaccination of the elderly population. Curr. Opin. Pharmacol. 41, 34–41 (2018).
Onodera, T. et al. Whole-virion influenza vaccine recalls an early burst of high-affinity memory b cell response through TLR signaling. J. Immunol. 196, 4172–4184 (2016).
Trombetta, C. M. & Montomoli, E. Influenza immunology evaluation and correlates of protection: a focus on vaccines. Expert Rev. Vaccines 15, 967–976 (2016).
Fonville, J. M. et al. Antibody landscapes after influenza virus infection or vaccination. Science 346, 996–1000 (2014).
Throsby, M. et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 3, e3942 (2008).
Corti, D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011).
Dreyfus, C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012).
Bournazos, S., DiLillo, D. J. & Ravetch, J. V. The role of Fc-FcγR interactions in IgG-mediated microbial neutralization. J. Exp. Med. 212, 1361–1369 (2015).
Pincetic, A. et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 15, 707–716 (2014).
Yamayoshi, S. et al. A broadly reactive human anti-hemagglutinin stem monoclonal antibody that inhibits influenza a virus particle release. EBioMedicine 17, 182–191 (2017).
Lee, J. et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat. Med. 22, 1456–1464 (2016).
McCarthy, K. R. et al. Memory B cells that cross-react with group 1 and group 2 influenza A viruses are abundant in adult human repertoires. Immunity 48, 174–184 e179 (2018).
Strauli, N. B. & Hernandez, R. D. Statistical inference of a convergent antibody repertoire response to influenza vaccine. Genome Med. 8, 60 (2016).
Laserson, U. et al. High-resolution antibody dynamics of vaccine-induced immune responses. Proc. Natl Acad. Sci. USA 111, 4928–4933 (2014).
Andrews, S. F. et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci. Transl. Med. 7, 316ra192 (2015).
Paules, C. I., Marston, H. D., Eisinger, R. W., Baltimore, D. & Fauci, A. S. The pathway to a universal influenza vaccine. Immunity 47, 599–603 (2017).
Ekiert, D. C. et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850 (2011).
Brandenburg, B. et al. Mechanisms of hemagglutinin targeted influenza virus neutralization. PLoS One 8, e80034 (2013).
DiLillo, D. J., Tan, G. S., Palese, P. & Ravetch, J. V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat. Med. 20, 143–151 (2014).
Steel, J. et al. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. MBio 1, e00018-10 (2010).
Yassine, H. M. et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 21, 1065–1070 (2015).
Lu, Y., Welsh, J. P. & Swartz, J. R. Production and stabilization of the trimeric influenza hemagglutinin stem domain for potentially broadly protective influenza vaccines. Proc. Natl Acad. Sci. USA 111, 125–130 (2014).
Impagliazzo, A. et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 349, 1301–1306 (2015).
Krammer, F., Pica, N., Hai, R., Margine, I. & Palese, P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J. Virol. 87, 6542–6550 (2013).
Hai, R. et al. Influenza viruses expressing chimeric hemagglutinins: globular head and stalk domains derived from different subtypes. J. Virol. 86, 5774–5781 (2012).
Nachbagauer, R. et al. Induction of broadly reactive anti-hemagglutinin stalk antibodies by an H5N1 vaccine in humans. J. Virol. 88, 13260–13268 (2014).
Wang, T. T. et al. Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes. Proc. Natl Acad. Sci. USA 107, 18979–18984 (2010).
Mallajosyula, V. V. et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc. Natl Acad. Sci. USA 111, E2514–2523 (2014).
Eggink, D., Goff, P. H. & Palese, P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J. Virol. 88, 699–704 (2014).
El Bakkouri, K. et al. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 186, 1022–1031 (2011).
Kolpe, A., Schepens, B., Fiers, W. & Saelens, X. M2-based influenza vaccines: recent advances and clinical potential. Expert Rev. Vaccines 16, 123–136 (2017).
Tao, W. et al. Consensus M2e peptide conjugated to gold nanoparticles confers protection against H1N1, H3N2 and H5N1 influenza A viruses. Antiviral Res. 141, 62–72 (2017).
Ekiert, D. C. et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489, 526–532 (2012).
Schmidt, A. G. et al. Viral receptor-binding site antibodies with diverse germline origins. Cell 161, 1026–1034 (2015).
Whittle, J. R. et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl Acad. Sci. USA 108, 14216–14221 (2011).
Strauch, E. M. et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 35, 667–671 (2017).
Kadam, R. U. & Wilson, I. A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing antibodies to influenza A hemagglutinin. Proc. Natl Acad. Sci. USA 115, 4240–4245 (2018).
Yewdell, J. W., Frank, E. & Gerhard, W. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J. Immunol. 126, 1814–1819 (1981).
Jameson, J., Cruz, J. & Ennis, F. A. Human cytotoxic T-lymphocyte repertoire to influenza A viruses. J. Virol. 72, 8682–8689 (1998).
Berthoud, T. K. et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin. Infect. Dis. 52, 1–7 (2011).
Stoloff, G. A. & Caparros-Wanderley, W. Synthetic multi-epitope peptides identified in silico induce protective immunity against multiple influenza serotypes. Eur. J. Immunol. 37, 2441–2449 (2007).
Lillie, P. J. et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 55, 19–25 (2012).
Pleguezuelos, O. et al. A synthetic influenza virus vaccine induces a cellular immune response that correlates with reduction in symptomatology and virus shedding in a randomized phase Ib live-virus challenge in humans. Clin. Vaccine Immunol. 22, 828–835 (2015).
Francis, T. Proceedings of the American philosophical society. Proc. Am. Philos. Soc. 104, 572–578 (1960).
Gostic, K. M., Ambrose, M., Worobey, M. & Lloyd-Smith, J. O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 354, 722–726 (2016).
Bodewes, R. et al. Recurring influenza B virus infections in seals. Emerg. Infect. Dis. 19, 511–512 (2013).
Yamayoshi, S., Watanabe, M., Goto, H. & Kawaoka, Y. Identification of a novel viral protein expressed from the PB2 segment of influenza A virus. J. Virol. 90, 444–456 (2016).
Wu, Y., Wu, Y., Tefsen, B., Shi, Y. & Gao, G. F. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 22, 183–191 (2014).
Wilson, I. A., Skehel, J. J. & Wiley, D. C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3A resolution. Nature 289, 366–373 (1981).
Air, G. M. Influenza neuraminidase. Influenza Other Respir. Viruses 6, 245–256 (2012).
Palese, P., Tobita, K., Ueda, M. & Compans, R. W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61, 397–410 (1974).
Matrosovich, M. N., Matrosovich, T. Y., Gray, T., Roberts, N. A. & Klenk, H. D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 78, 12665–12667 (2004).
Reid, A. H., Taubenberger, J. K. & Fanning, T. G. Evidence of an absence: the genetic origins of the 1918 pandemic influenza virus. Nat. Rev. Microbiol. 2, 909–914 (2004).
Watanabe, T. et al. Circulating avian influenza viruses closely related to the 1918 virus have pandemic potential. Cell Host Microbe 15, 692–705 (2014).
Viboud, C. et al. Global mortality impact of the 1957–1959 influenza pandemic. J. Infect. Dis. 213, 738–745 (2016).
Fraser, C. et al. Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324, 1557–1561 (2009).
Shinde, V. et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005-2009. N. Engl. J. Med. 360, 2616–2625 (2009).
Acknowledgements
We thank S. Watson for editing the manuscript. Our work described in this review was supported by the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from the Japan Agency for Medical Research and Development (AMED) (JP18fm0108006), by Leading Advanced Projects for medical innovation (LEAP) from AMED (JP18am001007), by Grants-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT) of Japan (16H06429, 16K21723, and 16H06434), by JSPS KAKENHI Grant Number 18K07141, and by the Center for Research on Influenza Pathogenesis (CRIP) funded by National Institute of Allergy and Infectious Disease contract HHSN272201400008C.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Y.K. has received speaker’s honoraria from Toyama Chemical and Astellas Inc.; has received grant support from Chugai Pharmaceuticals, Daiichi Sankyo Pharmaceutical, Toyama Chemical, Tauns Laboratories, Inc., Otsuka Pharmaceutical Co., Ltd., and Denka Seiken Co., Ltd.; and is a co-founder of FluGen. S.Y. has no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yamayoshi, S., Kawaoka, Y. Current and future influenza vaccines. Nat Med 25, 212–220 (2019). https://doi.org/10.1038/s41591-018-0340-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-018-0340-z
This article is cited by
-
Impact of sex on humoral immunity with live influenza B virus vaccines in mice
npj Vaccines (2024)
-
Phillyrin ameliorates influenza a virus-induced pulmonary inflammation by antagonizing CXCR2 and inhibiting NLRP3 inflammasome activation
Virology Journal (2023)
-
Influenza vaccine: a review on current scenario and future prospects
Journal of Genetic Engineering and Biotechnology (2023)
-
Co-evolution of immunity and seasonal influenza viruses
Nature Reviews Microbiology (2023)
-
Safety and immunogenicity of a phase 1/2 randomized clinical trial of a quadrivalent, mRNA-based seasonal influenza vaccine (mRNA-1010) in healthy adults: interim analysis
Nature Communications (2023)
