
Abstract
Prophylactic vaccination of rhesus macaques with rhesus cytomegalovirus (RhCMV) vectors expressing simian immunodeficiency virus (SIV) antigens (RhCMV/SIV) elicits immune responses that stringently control highly pathogenic SIV infection, with subsequent apparent clearance of the infection, in ~50% of vaccinees. In contrast, here, we show that therapeutic RhCMV/SIV vaccination of rhesus macaques previously infected with SIV and given continuous combination antiretroviral therapy (cART) beginning 4–9 d post-SIV infection does not mediate measurable SIV reservoir clearance during over 600 d of follow-up on cART relative to RhCMV/control vaccination. However, none of the six animals started on cART on day four or five, across both RhCMV/SIV- and RhCMV/control-vaccinated groups, those rhesus macaques with SIV reservoirs most closely resembling those of prophylactically RhCMV/SIV-vaccinated and protected animals early in their course, showed post-cART viral rebound with up to nine months of follow-up. Moreover, at necropsy, these rhesus macaques showed little to no evidence of replication-competent SIV. These results suggest that the early SIV reservoir is limited in durability and that effective blockade of viral replication and spread in this critical time window by either pharmacologic or immunologic suppression may result in reduction, and potentially loss, of rebound-competent virus over a period of ~two years.
Similar content being viewed by others


Main
Infection with the AIDS-causing human immunodeficiency virus (HIV) and SIV is associated with establishment of viral reservoirs, including latently infected cells and virus in immunologically privileged tissue sites, that are highly, if not completely, resistant to clearance by cART or any natural immunologic mechanism1,2,3,4,5. The defining characteristic of these reservoirs is that they lead to resumption of progressive HIV or SIV infection when any highly effective pharmacologic (cART) or immunologic (elite control) impediment to viral replication and spread is disabled, even after years of suppressed viral replication, and therefore constitute the major barrier to cure of HIV/SIV infection. Although the effectiveness of short-duration postexposure prophylaxis (PEP) with antiretroviral therapy indicates that HIV/SIV reservoir establishment is not immediate, this window of susceptibility to PEP has been considered to close in 2–3 d after virus exposure6,7, and it has been thought that after that time, certainly once infection is spreading systemically, an essentially permanent viral reservoir is established. Indeed, rhesus macaques treated with cART three days after intrarectal SIV challenge, before development of measurable viremia, showed resumption of progressive infection when cART was discontinued six months later8. Although systematic study of initiating cART during documented hyperacute HIV infection is often complicated by difficulty in precisely defining the exact timing of cART initiation relative to HIV exposure, rebound viremia has eventually occurred when cART was discontinued in virtually all such cases (although sometimes after long aviremic periods), consistent with early establishment of a persistent viral reservoir9,10,11,12,13.
Given the persistence of these HIV/SIV reservoirs and their resistance to immune elimination, it was therefore surprising that prophylactic administration of a strain 68-1 RhCMV/SIV vaccine (that is, vaccination before SIV challenge) resulted in stringent postacquisition control and eventual apparent clearance of highly pathogenic SIVmac239 infection in ~50% of vaccinated rhesus macaques14,15,16. This vaccine induces cellular, but not antibody, responses against SIV and the observed protective effects are thought to be mediated by SIV-specific, effector memory-type T cells (probably CD8+ T cells targeting unconventionally restricted SIV epitopes17,18) that intercept the infection in its early stages. Detailed virologic analysis of protected rhesus macaques indicates that control of viral spread occurs after dissemination of infection from the portal of viral entry, usually after documentation of transient plasma viremia14,15. These RhCMV/SIV vaccine-protected rhesus macaques harbor replication-competent virus in multiple tissues for weeks to months, but virologic and immunologic evidence of infection wanes over time, and after 1–3 yr, these once unequivocally SIV-infected rhesus macaques can no longer be distinguished from rhesus macaques that were vaccinated, but never challenged, even by extensive virologic analysis at necropsy14,15.
Although the immunologic mechanism(s) responsible for the RhCMV/SIV vaccine-mediated control and subsequent clearance of SIV remains to be precisely defined, the vaccine-elicited restriction of SIV replication and clearance of an existing AIDS virus infection is unprecedented, and suggests that either the permanent viral reservoir is susceptible to active elimination by these unusual immune responses, or the seeding of the permanent viral reservoir takes longer than previously thought, with both options having major implications for HIV prevention and cure. To distinguish between these possibilities and to determine whether RhCMV/SIV vaccine-elicited responses could clear a pre-established SIV reservoir, we infected rhesus macaques with SIVmac239 and then initiated cART at early times (4–9 d) postinfection, with treatment initiation triggered based on the earliest evidence of a systemic host response to infection19,20. Our goal was to first establish a range of reservoir sizes that approximated the extent of the initial SIV reservoir typical of SIV-infected rhesus macaques protected by prophylactic RhCMV/SIV vaccination, and then, keeping the infected rhesus macaques on continuous cART, determine whether responses elicited by a subsequent therapeutic vaccination with the same RhCMV/SIV vectors could reduce or eliminate this reservoir. We found that while RhCMV/SIV-elicited, SIV-specific immune responses had no demonstrable impact on the pre-established SIV reservoir relative to control responses elicited by RhCMV vectors expressing a non-SIV antigen insert, 6 rhesus macaques, including animals from both vaccine groups, started on cART before 6 d postinfection (dpi) and maintained on cART for ~600 d, did not manifest rebound viremia during up to 9 months of post-cART follow-up. These data suggest that the long-term SIV reservoir is not fully established until ~6 dpi, and that if SIV replication and spread are effectively suppressed before this critical time point, and this suppression is maintained for ~2 yr, the SIV reservoir may decline to the extent that after treatment discontinuation viral recrudescence may be substantially delayed and potentially prevented.
Results
SIV reservoir establishment and vaccination
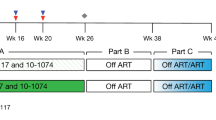
About 50% of RhCMV/SIV-vaccinated rhesus macaques stringently control SIV infection in the first 7–14 d after challenge, leaving the protected rhesus macaques with a widely tissue-distributed, but low-level and non-spreading (largely aviremic) infection14,15,16 (Supplementary Fig. 1). Replication-competent SIV is detectable in RhCMV/SIV-protected rhesus macaques in the first few months postchallenge14,15 (Supplementary Fig. 1) but, with few exceptions, the ability to detect persisting SIV by all virologic or immunologic criteria declines over the subsequent 12 months until, by 1–3 yr postchallenge, the protected, once-infected rhesus macaques can no longer be distinguished from vaccinated, but never challenged animals14,15. Here, we sought to determine the extent to which immune responses elicited by therapeutic RhCMV/SIV vaccination can clear a previously established, cART-suppressed SIV reservoir that approximates the level of viral spread observed in such protected, prophylactically strain 68-1 RhCMV/SIV-vaccinated rhesus macaques (Fig. 1a).

a, Schematic representation of the study protocol showing SIVmac239X infection, cART initiation 4–9 dpi, RhCMV/SIV or control vector vaccination at 91, 175, and 399 dpi, and cART withdrawal at 611 dpi. b, The mean (+ s.e.m.) frequencies of CD169 expression on CD14+ monocytes in blood of rhesus macaques stratified by time of cART initiation; that is, 4–5 dpi (blue; n = 6), 6 dpi (red; n = 13), 7 dpi (green; n = 10), or 8–9 dpi (purple; n = 4). c, Mean (+ s.e.m.) plasma viral load (pvl) profiles (left panel) and pvl area under the curve (AUC) between 0 and 84 dpi (right panel) in rhesus macaques stratified by time of cART initiation. Pvl profiles and AUC of unvaccinated, comparably SIVmac239-infected rhesus macaques that initiated cART at 12 dpi (orange; n = 12) and 42 dpi (black; n = 18) were included for comparison. Overall difference was determined by two-sided Kruskal–Wallis test and where that was significant (P values ≤0.05), two-sided Wilcoxon rank-sum test (WRS) was used to determine the significance of pair wise differences in the AUC (WRS P values shown). Plots show jittered points with a box from first to third quartiles and a line as the median, with whiskers extending to the farthest data point within 1.5 × interquartile range above and below the box, respectively. d, Longitudinal quantification of SIV RNA (left) and DNA (right) in PBMCs (mean + s.e.m.; copies per 106 cell equivalents) between 0 and 84 dpi in rhesus macaques stratified by time of cART initiation. Threshold sensitivity varied as a function of the number of cells available for analysis; for graphing consistency values are plotted with a common nominal sensitivity threshold of 1 copy of SIV RNA or DNA per 108 cell equivalents, indicated by a dotted gray line. e,f, Quantification of SIV RNA (left) and DNA (right) in lymph nodes (copies per 106 cell equivalents) at 3 and 70 d post-cART initiation. Each data point represents a single determination from an individual rhesus macaque. c–f, Sample sizes are as shown in b, black (historical control) n = 18 and orange (day 12) n = 12. In c, e, and f, the Kruskal–Wallis test was used to determine the significance of differences between day-of-cART initiation groups and when that test was significant (P values ≤0.05); the WRS test was used to determine the significance of pair wise differences among the cART treatment groups (WRS P values shown). Plots show jittered points with a box from first to third quartiles and a line as the median, with whiskers extending to the farthest data point within 1.5 × interquartile range above and below the box, respectively. The dotted gray line indicates a threshold of 0.6 copies of SIV RNA or DNA per 106 cell equivalents. Days 7, 8, and 9 were pooled for these comparisons due to the small sample size at days 8 and 9 (n = 4 combined), with the overall n the same as in panels a and b. TDF, tenofovir disoproxil fumarate; FTC, emtricitabine (2',3'-dideoxy-5-fluoro-3'-thiacytidine); DTG, dolutegravir.
Since SIV levels increase very rapidly in primary SIV infection, ~1-log per day increase in plasma viral load (pvl)21, and since peak viremia in protected, prophylactically RhCMV/SIV-vaccinated rhesus macaques is detected in most animals, but quite low (median peak pvl = 2,600 copies ml−1; Supplementary Fig. 1), early cART initiation is required to recapitulate the limited extent of viral spread characteristic of prophylactically vaccinated infected rhesus macaques that go on to control and clear their infections. We therefore elected to initiate cART22 at the first sign of a systemic take of SIV infection after intravenous challenge with two focus-forming units of SIVmac239X (an isogenic sequence-tagged version of the highly pathogenic SIVmac239 clone)23. To this end, we used rapid flow cytometric detection of monocyte activation (assessed as interferon-triggered upregulated expression of CD169 on ≥20% of CD14+ monocytes19,20) as an early systemic measure of innate immune detection of infection (Supplementary Fig. 2), allowing for cART initiation within 8 h of collection of the first above-threshold blood sample.
As shown in Fig. 1b, the onset of monocyte activation was heterogeneous among 33 SIVmac239X-challenged rhesus macaques with 6 showing above-threshold responses at 4 or 5 dpi, 13 at 6 dpi, 10 at 7 dpi, and 4 at 8 or 9 dpi; in each rhesus macaque, cART was initiated on the day of the first above-threshold monocyte response. This approach did not result in the same extent of viral spread across the 33 rhesus macaques, but rather resulted in varying levels of infection that reflected the day of cART initiation. The later cART was initiated, the greater the peak pvl, the pvl area under the curve (AUC) from 0–84 dpi, and the peak cell-associated SIV RNA and DNA in peripheral blood mononuclear cells (PBMCs) and lymph nodes, all with ~1-log increase in the degree of infection for each extra day of infection before cART initiation (Fig. 1c–f). Of note, all rhesus macaques were initially viremic (with at least 4 above threshold pvl measurements), and all of these infected rhesus macaques became aviremic (<30 SIV RNA copies ml−1) by 42 dpi. With few exceptions, pvls remained at <30 SIV RNA copies ml−1 thereafter. Once stable suppression was established, cell-associated SIV RNA in PBMCs and lymph nodes was reduced to near or below threshold levels. In contrast, cell-associated SIV DNA in these same sites declined only modestly (~1-log), and therefore continued to show the same step-wise differences in levels related to the day of cART initiation (Fig. 1d–f), consistent with a step-wise increase of extent of the overall SIV reservoir. For comparison, we report viral quantification data from 2 additional rhesus macaque cohorts given the same SIVmac239 challenge, but placed on cART at 12 dpi (the time of peak pvl in primary infection with this challenge route) and (for blood measurements only) 42 dpi. Of note, suppression of plasma viremia with the same cART regimen took longer (cART initiated 12 dpi) or was less complete (cART initiated 42 dpi) in these rhesus macaques, and cell-associated viral reservoirs were higher than for rhesus macaques placed on cART at earlier time points (Fig. 1c–f).
The 33 rhesus macaques described above (all naturally RhCMV-infected before study assignment) were assigned to balanced groups based on pvl AUC through 84 dpi, and then were vaccinated with a strain 68-1 RhCMV-vectored vaccine containing either SIV inserts (gag/pol/retanef/env14,15) or a control insert (Mycobacterium tuberculosis Ag85B/ESAT614), at 91, 175, and 399 dpi, with continuous cART administration (Fig. 1a). Before vaccination, rhesus macaques in both the RhCMV/SIV- and control vector-vaccinated groups showed induction of CD4+ and CD8+ T cell responses to the SIV insert antigens (Gag, Rev/Tat/Nef, Pol, and Env) and the non-insert SIV antigen Vif, reflecting priming of these SIV-specific responses by the SIV infection. However, after vaccination only the RhCMV/SIV-vaccinated rhesus macaques showed boosting of the SIV insert antigen-specific response, with no effect on responses to the non-insert SIV antigen (Vif), and the de novo induction of the unconventional major histocompatibility complex (MHC)-E- and MHC-II-restricted supertope-specific responses (Gag53211-222, Gag69276-284, Gag73290-301, Gag120482-490), respectively, that are characteristic of strain 68.1 RhCMV/SIV vaccination17,18 (Fig. 2a,b). Vaccination was not associated with measurable pvl ‘blips’ (Fig. 2c) and RhCMV/SIV vector vaccination did not result in an appreciable decrease in cell-associated SIV RNA or DNA compared to RhCMV/control vector vaccination in PBMCs, lymph node biopsy samples, or small intestinal mucosal biopsy samples (Fig. 2d–f).

a, Mean (+ s.e.m.) frequencies of peripheral blood memory CD4+ (top panels) and CD8+ (bottom panels) T cells specific for SIV proteins (Gag, Rev/Tat/Nef, Env, and Pol) expressed by RhCMV/SIV vectors, and for SIVvif (not included in the RhCMV/SIV vectors) in RhCMV/SIV-vaccinated rhesus macaques (n = 17) versus RhCMV/control vector-vaccinated rhesus macaques (n = 16). These response frequencies were determined by intracellular expression of TNF-α and/or IFN-γ after stimulation with mixes of consecutive, overlapping SIV 15mer peptides for each SIV protein (Gag, Env, Pol, Vif) or protein combination (Rev/Tat/Nef), as described in the Methods section, with the total response to the SIV proteins expressed by RhCMV/SIV vaccine reflecting the sum of the Gag, Rev/Tat/Nef, Env, and Pol responses. RhCMV/SIV or RhCMV/control vaccine administration is indicated by arrows. b, Mean (+ s.e.m.) frequencies of SIV Gag supertope responses (Gag53211-222 (AADWDLQHPQP, MHC-II-restricted); Gag69276-284 (RMYNPTNIL, MHC-E-restricted); Gag73290-301 (PKEPFQSYVDRF, MHC-II-restricted); Gag120482-490 (EKQRESREK, MHC-E-restricted)17,18) within peripheral blood CD8+ memory T cell compartment of RhCMV/SIV vector-vaccinated rhesus macaques (left panel; n = 17) and control RhCMV vector-vaccinated rhesus macaques (right panel; n = 16). c, Pvl (mean + s.e.m.) profiles of RhCMV/SIV vector-vaccinated (n = 17) and control RhCMV vector-vaccinated (n = 16) rhesus macaques on cART measured at a threshold of 30 RNA copies per ml from 0 to 175 dpi and 1 RNA copy per ml from 189 to 609 dpi (same groups as in a). AUC calculated based on LOD = 30 for all time points. d–f, Longitudinal quantification of cell-associated SIV DNA (left) and SIV RNA (right) (mean + s.e.m.; copies per 106 cell equivalents) in the designated tissues from RhCMV/SIV vector-vaccinated rhesus macaques (n = 17) versus the RhCMV/control (n = 16) vector-vaccinated rhesus macaques (RhCMV vaccination indicated by arrows). As described in Fig. 1 legend, values are plotted with a common nominal sensitivity threshold of 1 copy of SIV RNA or DNA per 108 cell equivalents. In a and c–f, the two-sided WRS test was used to determine the significance of differences in the AUC (P values shown). CMV, cytomegalovirus; LOD, limit of detection.
Rebound viremia after cART discontinuation is related to time of cART initiation, but not to the type of vaccine received
At 611 dpi, corresponding to 603–607 d of cART administration and 520 d of exposure to RhCMV/SIV- or RhCMV/control-elicited responses, cART was discontinued, and the study rhesus macaques monitored for rebound viremia. There was no significant difference in either the frequency of rebound or time to measurable rebound viremia between the two vaccine groups (Fig. 3a,b), consistent with the RhCMV/SIV vaccination not conferring any benefit in the present therapeutic vaccination regimen, either by reducing the SIV reservoir before cART release, or by providing postrebound viral control after cART release. Strikingly, however, 7 rhesus macaques (4 in the RhCMV/control-vaccinated group and 3 in the RhCMV/SIV-vaccinated group) did not show evidence of sustained off-cART rebound viremia through 6 months of post-cART follow-up (1 of 7 subsequently rebounded, see below), and an additional 5 rhesus macaques (2 in the RhCMV/control-vaccinated group and 3 in the RhCMV/SIV-vaccinated group) showed delayed rebound (≥56 d post-cART release; Fig. 3b) within the 6 months of formal follow-up. All but 1 of these 12 rhesus macaques were administered cART starting at or before 6 dpi.

a, Kaplan–Meier analysis of SIV rebound kinetics in RhCMV/SIV vector-vaccinated (n = 17) and control RhCMV vector-vaccinated (n = 16) rhesus macaques post-cART cessation. The significance of the difference between groups was determined using a two-sided log-rank test (P value shown). b, Pvl profiles after cART release stratified by vaccine group measured at a threshold of 1 RNA copy per ml. Early rebound (n = 21; black) was defined as sustained rebound viremia occurring <40 d after cART release, late rebound (n = 5; blue) defined as sustained viremia >40 d after cART release, and no rebound (n = 7; pink) defined as no sustained viremia through 180 d after cART release. c, Pvl profiles after cART release of study rhesus macaques stratified by time of cART initiation and measured at a threshold of 1 RNA copy per ml. d, Kaplan–Meier analysis of SIV rebound kinetics by time of cART initiation. Significance of difference between groups was determined using a two-sided log-rank test (P values shown). e, Spearman rank correlation between the AUC of pvl from 0 to 84 dpi versus time to virus rebound (two-sided rank correlation t-test; P value shown). Rhesus macaques taken to necropsy after 205 dpi are plotted at the day of necropsy. In c–e, rhesus macaque groups and sample sizes are the same as in Fig. 1b.
Given the absence of any apparent protective effect of RhCMV/SIV vaccination on the frequency of, or time to, rebound viremia, and the suggestion of an effect of time of cART initiation on these parameters, we reanalyzed our data based on time of cART initiation rather than vaccine group. In addition to the finding that the rhesus macaques that did not show rebound viremia were those in which cART was initiated earliest, among the remaining rhesus macaques that did show rebound viremia, the time to measurable rebound viremia was inversely related to the time of cART initiation after infection (Fig. 3c,d) and correlated with the extent of viral replication before initiation of cART, as reflected by the plasma SIV RNA viremia AUC measurement through 84 dpi (used as an index of cumulative viral infection; Fig. 3e). Time to rebound was also inversely related to other viral spread indices, including peak pvl and peak PBMC and lymph node cell-associated SIV viral RNA (vRNA) and SIV viral DNA (vDNA) levels (Supplementary Fig. 3). As indicated above, all rhesus macaques were viremic for ≥4 consecutive days before cART-mediated viral suppression, indicating that all animals had a spreading SIV infection before initiating cART. In keeping with this, all study rhesus macaques manifested CD4+ and CD8+ T cell responses to the non-vaccine SIVvif antigen on infection, and the magnitude and duration of these responses were similar across the rhesus macaque groups with different days of cART initiation and between rhesus macaques that manifested early versus late versus no SIV rebound within six months after cART cessation (Supplementary Fig. 4).
The lack of SIV rebound in the rhesus macaques with early cART initiation was not due to the ability of qualitatively superior SIV-specific CD8+ T cell responses to suppress viral replication, as treatment of 3 of these post-cART non-rebounders with a depleting anti-CD8α monoclonal antibody24,25 ~185 d after cART discontinuation (Fig. 4a) did not result in detectable SIV viremia. In contrast, parallel CD8+ cell depletion of 3 rhesus macaques that showed rebound viremia but established post-cART elite control (pvl <10,000 copies ml−1) resulted in an immediate 2–3-log increase in viremia that partially or completely returned back to predepletion levels in concert with partial recovery of the circulating CD8+ T cell population (Fig. 4a).

a, Effect of CD8+ lymphocyte depletion on absolute CD8+ T cell counts in blood (top panel) and pvl (bottom panel) in three rhesus macaques with no viral rebound (red) versus a control group of three rhesus macaques with post-cART rebound but subsequent virologic control (black). Rhesus macaques received anti-CD8α M-T807R1 monoclonal antibody at 10, 5, 5, and 5 mg per kg body weight on days 0, 3, 7, and 10. Note that CD8+ cell depletion increased plasma viremia by ~3-logs in the control group, but had no effect on pvl in the non-rebounders. b, Assessment of residual replication-competent, cell-associated SIV by adoptive transfer of lymph node cells (6 × 106 to 1 × 108) from post-cART non-rebounders to SIV-naïve rhesus macaques with the transfer of SIV infection to the recipient rhesus macaques delineated by sustained, above threshold (15 RNA copies ml−1) pvl. Cell transfers before 175 dpi resulted in rapid onset of SIV infection in the recipient rhesus macaques, but no SIV infection was observed in rhesus macaques receiving cells 720 dpi (109 d after cART cessation). c, Assessment of residual replication-competent, cell-associated SIV by adoptive transfer of lymph node cells from 2 rhesus macaques with post-cART rebound <180 d after cART release. Cell transfers from rhesus macaque 27812 at 11 dpi resulted in rapid onset of SIV infection in the recipient rhesus macaque, but no SIV infection was observed in rhesus macaques receiving cells from either rhesus macaque at 720 dpi. d, Assessment of residual replication-competent, cell-associated SIV by adoptive transfer of lymph node cells from 720 dpi from a rhesus macaque with post-cART rebound at necropsy (206 d post-cART cessation) to SIV-naive rhesus macaques, showing no SIV infection in recipient rhesus macaques. RM, rhesus macaque.
Viral clearance in late and non-rebounding rhesus macaques
To better understand the virologic status of rhesus macaques with delayed rebound or no rebound in the six months after cART cessation, we assessed the presence of replication-competent SIV in lymph nodes of these rhesus macaques at different time points using the adoptive transfer approach15,26,27 (Supplementary Fig. 1). Adoptive transfer of 23 × 106 lymph node mononuclear cells (LNMCs) from rhesus macaque 28153 (day 4 cART initiation; non-rebounder) at 7 dpi (Fig. 4b), or only 6 × 106 LNMCs from rhesus macaque 27812 (day 7 cART initiation; late rebounder) at 11 dpi (Fig. 4c), when the animals had only been on cART for 3–4 d, readily transmitted virologically typical SIV infection to naïve rhesus macaques. Similarly, adoptive transfer of 30 × 106 LNMCs from rhesus macaque 24458 and rhesus macaque 27380 (both day 4 cART initiation; non-rebounders), at 175 dpi, while still on cART and when they had been aviremic for ~150 d, also readily transmitted typical SIV infection (Fig. 4b), indicating the presence of residual replication-competent virus in these rhesus macaques in the first 6 months after early cART initiation. The presence of this transferrable replication-competent virus after ~ 6 months of cART, which would be expected to give rise to progressive infection and rebound viremia had cART been discontinued after only 6 months in our rhesus macaques, is thus consistent with previous results showing SIVmac251 rebound 6 months following cART initiation at 3 dpi8. Taken together, these results indicate that the SIV reservoirs established by 3–5 dpi are able to persist for at least 6 months.
However, at 720 dpi, more than 100 d after cART discontinuation, adoptive transfers of 60–100 × 106 LNMCs from the 7 initially (through 6 months) non-rebounding rhesus macaques did not transfer SIV infection to naïve recipients (Fig. 4b,d and Supplementary Fig. 5), suggesting that cells containing transferable virus were not present in these cell preparations. For 6 of these 7 non-rebounding rhesus macaques (24458, 27380, 28153, 23187, 28444, 28214), followed for an additional 97–147 d after adoptive transfers at 720 dpi had failed to transmit infection (until euthanasia and necropsy), there was no evidence of sustained rebound viremia. A negative adoptive transfer result, however, does not preclude the presence of replication-competent virus elsewhere in the animal as 1 of these 7 rhesus macaques, 27706, the only day 6 cART initiation non-rebounder, showed late viral rebound at its 817 dpi necropsy, 206 d after cART release (Fig. 4d). In addition, two other delayed rebounders (27812, 23813) showed a negative adoptive transfer result before subsequent overt viral rebound (Fig. 4c). Together, these observations of late rebounding and non-rebounding rhesus macaques are consistent with residual replication-competent virus being eliminated over time in these animals, or, alternatively, reduced over time to such an extent that replication-competent virus becomes inhomogeneously distributed across the various lymphoid tissues, but is still able to reactivate and rekindle systemic infection. These results are reminiscent of dramatically delayed and unpredictable stochastic viral recrudescence seen in certain clinical circumstances9,10,28.
Necropsy analyses
Rhesus macaques that failed to manifest SIV rebound in the 6 months following cART cessation (n = 7) were euthanized and taken to necropsy from 206–256 d after stopping cART, and extensive analyses were performed on necropsy tissues with 118–122 tissue samples tested per rhesus macaque, including tissue sites known to harbor SIV. As noted above, one of these rhesus macaques (27706) showed plasma viremia at necropsy indicative of very recent rebound (Fig. 4d), whereas the other six did not. Using sensitive and specific nested qPCR/qPCR with reverse transcription testing15,29, SIVgag DNA or RNA target sequences were either not detected, or detected at only very low levels in the tissues analyzed from the six non-rebounding rhesus macaques, with vRNA detected less frequently than vDNA (Fig. 5a). In contrast, rhesus macaque 27706 showed widely and uniformly disseminated vDNA and ~3-log higher levels of vRNA, broadly similar to overtly SIV-infected controls (Fig. 5b–d).

a–d, Quantification of cell-associated SIV DNA (blue) and SIV RNA (red) (copies per 106 cell equivalents) in the designated tissue samples of all 6 rhesus macaques with no post-cART rebound viremia through necropsy, the rhesus macaques with virus rebound at necropsy (pvl = 75,000 RNA copies ml−1), a representative post-cART elite controller (pvl = 270 RNA copies ml−1), and a viremic progressor (pvl = 350,000 RNA copies ml−1). Each symbol represents a single determination from a specimen of the designated tissue type (closed circles reflect DNA or RNA measurements above threshold; open circles DNA or RNA measurements below the assay threshold (see Methods)). Threshold values for individual samples varied as a function of the amount of tissue available and analyzed, and ranged from 0.7 to 18 copies per 106 cell equivalents for DNA (mean 5.5 and median 2.8 copies per 106 cell equivalents, respectively) and from 0.6 to 148 copies per 106 cell equivalents for RNA (mean 26 and median 4.1 copies per 106 cell equivalents, respectively). Also shown in green are the frequencies of SIV+ cultures (2.5 × 105 cells per culture) from the designated tissues after coculture with CEMx174 for 17 d. e, Detection of replication-competent SIV after 12 h PMA/ionomycin stimulation of sorted CD4+ T cells from lymph node or spleen (5 × 105 sorted CD4+ cells per well cocultured with CEMx174 for 21 d; the total number of cells analyzed from each rhesus macaque is shown in the figure) from all 6 of the post-cART non-rebounders, the rhesus macaques with virus rebound at necropsy, and a representative post-cART elite controller. f, Assessment of residual replication-competent, cell-associated SIV in all 6 post-cART non-rebounders and, as a control, an elite controller by adoptive transfer of 1 × 108 hematolymphoid cells that were positive for SIV DNA at necropsy (including cells from lymph node, spleen, and liver; see Table 1) to SIV-naïve rhesus macaques. Cell transfer from the elite controller rhesus macaque induced rapid onset of SIV infection in the recipient rhesus macaques, but no SIV infection was observed in rhesus macaques receiving cells from the six rhesus macaques with no post-cART virus rebound. BM, bone marrow; LN, lymph node; GI, gastrointestinal; CNS, central nervous system; pos., positive; VOA, viral outgrowth assay.
In the 6 non-rebounding rhesus macaques, testing for replication-competent virus by extensive cocultures of 33–55 × 106 total cells from selected necropsy tissues (Fig. 5a) or by phorbol myristate acetate (PMA)/ionomycin stimulation of 25–55 × 106 sort-purified CD4+ T cells from lymph node or spleen (Fig. 5e) did not recover replication-competent virus except for extremely rare, sporadic positive cultures from peripheral lymph nodes (9 of 2,520 separate cultures, with 2.5 × 105 cells input per culture; 0.3%). In contrast, replication-competent virus was recovered from 980 of 1,297 (76%; P <0.0001) cultures of tissues taken at necropsy from the overtly infected animals (Fig. 5b–d), and 100% of cultures of PMA/ionomycin-stimulated CD4+ T cells (Fig. 5e). Finally, adoptive transfer of up to 100 × 106 total cells from low-level vDNA+ tissues from the non-rebounding rhesus macaques did not transmit infection to naïve rhesus macaques (Fig. 5f and Table 1). Taken together, these findings are consistent with the interpretation that some or all of the rare, low-level vRNA and vDNA signals detected may have represented either assay false positives or, alternatively, represented nucleic acid assay true positives, but reflecting viral sequences derived from non-replication-competent proviruses30,31.
Discussion
The ability of RhCMV/SIV vaccination before SIV challenge to mediate postacquisition control and eventual apparent clearance of infection that had already spread to multiple tissues14,15 suggested that such vaccines might have activity in therapeutic vaccination settings. To evaluate the potential utility of therapeutic vaccination with RhCMV/SIV, we infected animals with SIVmac239 and initiated cART at the earliest systemic sign of a take of the infection (4–9 dpi), a strategy intended to approximate the extent of viral seeding seen in prophylactically RhCMV/SIV-vaccinated rhesus macaques that went on to control and apparently clear their infections. Once viral suppression was achieved, animals were vaccinated with RhCMV/SIV versus RhCMV/ control vectors, cART was discontinued after over 600 d of continuous treatment, and viral rebound dynamics were assessed. In this experimental protocol, therapeutic RhCMV/SIV vaccination did not appear to confer any advantage over control vaccination with respect to the frequency, kinetics, or extent of rebound viremia. However, these parameters were profoundly influenced by the timing of cART initiation. Remarkably, independent of the vaccine received, all six rhesus macaques placed on cART within five days of SIVmac239 infection, though unequivocally infected in the initial weeks to months after challenge, failed to rebound when cART was discontinued, with little or no evidence of replication-competent virus detected by extensive tissue analysis at necropsy, approximately eight months after cART discontinuation. Six other rhesus macaques that started cART at 6 dpi (n = 5) or 7 dpi (n = 1) showed delayed viral rebound after cART cessation, including 1 rhesus macaque that had rebounded just days before necropsy, after ~8 aviremic months off-cART, consistent with stochastic reactivation of a profoundly reduced, highly quiescent replication-competent SIV reservoir.
This lack of SIV rebound in all of the rhesus macaques placed on cART at 4 or 5 dpi and the delayed SIV rebound in many of the rhesus macaques given cART starting on day 6 complicates interpretation of the apparent lack of RhCMV/SIV vaccine efficacy in this study, as viral reservoirs that most closely resemble the reservoirs that clear in prophylactically RhCMV/SIV-vaccinated and protected rhesus macaques appear to clear or reduce with extended cART regardless of whether an SIV-targeted or control vaccine was received. The exponentially larger reservoirs of rhesus macaques started on cART at day seven or later were not affected by RhCMV/SIV-elicited, SIV-specific T cell responses, indicating that these responses either lacked the necessary effector activity or were unable to access and/or recognize the cells harboring rebound-competent SIV over the time period of cART used in this study. We also did not detect any evidence of the stringent control of SIV replication observed in prophylactic RhCMV/SIV vaccination14,15 in the 14 therapeutically RhCMV/SIV-vaccinated rhesus macaques that showed post-cART viral rebound in this study. Since all of these rhesus macaques would have established systemically distributed SIV reservoirs before suppression of viral replication (Fig. 1e), probably including SIV-infected cells located in immunologically privileged sanctuary sites such as B cell follicles3,4,32, the trajectory of SIV infection arising from post-cART viral reactivation is likely to be very different than that of primary infection. In the latter situation, an immediate immune intercept of the nascent infection might in fact prevent seeding of sanctuary sites; whereas in the former situation, such sites would have been seeded before establishment of the effector memory T cell responses. Failure of RhCMV/SIV vector-elicited responses to mediate stringent control off-cART might therefore be a function of limited effector T cell access to productively infected cells early after reactivation3,32.
In addition, while we did observe induction of the unconventionally targeted SIV-specific CD8+ T cell responses characteristic of strain 68-1 RhCMV/SIV-vaccinated rhesus macaques in the therapeutically RhCMV/SIV-vaccinated rhesus macaques, it is unclear whether these immune responses have the same in vivo effector potency as those elicited by the same vaccine used prophylactically in SIV-naïve rhesus macaques. In the therapeutic vaccination setting, the pre-existing SIV-specific immune responses arising from SIV infection, the innate immune activation resulting from this challenge and its persistence after cART suppression, and even the cART itself might modify the magnitude, function, or distribution of the RhCMV/SIV-elicited responses relative to vaccination of naïve rhesus macaques. As correlates of RhCMV/SIV-mediated protection in the prophylactic vaccination setting are definitively established by ongoing research, the presence or absence of these correlates will need to be evaluated in the therapeutic vaccine setting, with further development of therapeutic cytomegalovirus vectors for prevention of HIV/SIV outgrowth after cART release to depend on whether these correlates can be generated by RhCMV/SIV vectors in rhesus macaques with pre-existing cART-suppressed SIV infection.
Taken together, the results suggest that interruption of SIV spread in early primary infection by either cART (this study) or prechallenge-elicited RhCMV/SIV-elicited immune responses14,15,16 can prevent establishment of a long-term/permanent SIV reservoir, leaving behind a relatively short-lived SIV reservoir that can decline over the subsequent 12–20 months, either to the point of apparent effective clearance, or to the point that rebound viremia becomes profoundly delayed and stochastic. The results underscore the apparent heterogeneity of viral reservoirs, including a component that declines to, or nearly to extinction when viral spread is stringently blocked for 6–20 months, as well as elements that durably persist beyond 20 months. The quantitative and qualitative features that contribute to this heterogeneity remain to be defined, but quantitative increase of the reservoir above a critical threshold, as well as viral penetration of different anatomic and cellular compartments, including establishment of expanded clones harboring clonally integrated proviruses5,33,34,35,36,37,38, are potential factors that bear further scrutiny. It should also be explicitly noted that while the current study included a control group of rhesus macaques vaccinated with RhCMV containing a non-SIV antigen insert, for practical and logistical reasons, the study did not include an unvaccinated group that received cART only. Thus, although persistent RhCMV infection is ubiquitous in monkeys used for SIV cure studies, we cannot formally rule out the possibility that effects of the RhCMV vaccine vector super-infection, independent of the antigen-specific inserts, may have influenced the outcomes observed.
While anecdotal clinical reports suggest that, similar to the rhesus macaques receiving early treatment in this study, initiation of cART during the hyperacute phase of HIV infection can profoundly reduce viral reservoir establishment and dramatically delay viral rebound after treatment is interrupted9,10,12,13, such early initiation of cART is not relevant to the majority of HIV-infected individuals, who typically start treatment well into the chronic phase of infection. However, the conclusion that the earliest SIV reservoir can apparently decay may have important implications for prophylactic HIV/AIDS vaccine development, as it implies that an effective prophylactic vaccine need not completely prevent infection at the portal of entry, but rather can be efficacious (for example, result in an exposed individual remaining HIV− over the long term) as long as immunologic mechanisms are able to stringently arrest viral spread, potentially via both cytolytic and non-cytolytic mechanisms, before the establishment of longer-term/permanent reservoirs. Given that cellular immunity acts on infected cells, such early stringent viral arrest is the major, if not only, mechanism by which vaccine-elicited cellular immunity (in particular that induced by cytomegalovirus-based vectors) can mediate complete protection. However, it is probably necessary for the effectiveness of vaccine-elicited humoral immunity as well, as recent data in rhesus macaque studies suggest that even protective doses of potent passively administered broadly neutralizing antibodies fail to completely prevent target cell infection, but rather permit a limited initial infection, including at distal tissue sites, that decays or is cleared over approximately one week, preventing establishment of a fully disseminated systemic infection27,39. Thus, therapies aimed at eliminating HIV-infected cells or suppressing viral spread in early primary infection may have the potential to contribute to HIV prevention strategies.
On the other hand, in terms of HIV cure, it is sobering that despite substantial decay to levels not detectable by extensive sampling using sensitive methods, including adoptive transfer of large numbers of cells to a naïve host, replication-competent SIV was able to persist and initiate recrudescent systemic infection over nine months after cART discontinuation. Thus, an eradicative HIV/SIV cure may not be achievable with a 3- or even 4-log reduction in reservoir size, but rather might require elimination of every cell containing replication-competent virus.
Methods
Rhesus macaques
These experiments used a total of 84 purpose-bred male and female rhesus macaques (Macaca mulatta) of Indian genetic background with the approval of the Oregon National Primate Research Center’s Animal Care and Use Committee, under the standards of the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. These rhesus macaques were specific pathogen-free as defined by being free of cercopithicine herpesvirus 1, D-type simian retrovirus, simian T-lymphotrophic virus type 1, rhesus rhadinovirus, and M. tuberculosis. MHC-1 genotyping for common Mamu alleles such as Mamu-A*01/-A*02 and Mamu-B*08/-B*17 was performed by sequence-specific priming PCR, essentially as described previously40. The 84 rhesus macaques included 33 rhesus macaques (see Supplementary Table 1) that were intravenously inoculated with 2 TZM-bl assay focus-forming units of rhesus macaque PBMC-expanded SIVmac239X and placed on cART starting 4–9 dpi and vaccinated with RhCMV/SIV (n = 17) or control RhCMV vectors (n = 16) at 91, 175, and 399 dpi and maintained on cART for up to 610 dpi. Another 30 rhesus macaques (3–4 yr of age) challenged intravenously with the same dose of SIVmac23X and placed on cART 12 dpi (n = 12) or 42 dpi (n = 18) were used for virologic comparisons. cART consisted of a subcutaneous injection of 5.1 mg kg−1 d−1 tenofovir disoproxil, 40 mg kg−1 d−1 emtricitabine (FTC), and 2.5 mg kg−1 d−1 dolutegravir in a solution containing 15% (v/v) kleptose at pH 4.2, as previously described22. The 18 rhesus macaques that initiated cART on day 42 also received 600 mg darunavir twice daily boosted with 100 mg ritonavir twice daily (both given orally in food) for 96 d3. A total of 21 SIV-naïve rhesus macaques (4–15 yr of age) were used as RhCMV vector-naïve recipients in the adoptive transfer experiments. Selected rhesus macaques with post-cART viral control or no viral rebound after cART cessation were depleted of CD8+ lymphocytes by administration of the CD8α-depleting monoclonal antibody M-T807R1 (National Institutes of Health Nonhuman Primate Reagent Resource Program), which was injected subcutaneously (10 mg kg−1) at day 0, and intravenously (5 mg kg−1) at days 3, 7, and 1024,25. Adoptive transfer of mononuclear cells from SIV-infected rhesus macaques (on cART or after cART withdrawal) to allogeneic SIV-naïve rhesus macaques was used to detect the presence of cell-associated replication-competent virus in the donor rhesus macaques, as previously described15. This biologic viral detection assay was designed to reveal both productively and latently infected cells in the donor, the latter via reactivation in the recipient rhesus macaques due to the allogeneic stimulation of the donor CD4+ T cells in recipient, with reciprocal allogeneic activation of recipient CD4+ T cells by donor cells facilitating their infection (note: SIV infection is transferred from donor to recipient before the donor cells are eliminated from the recipient by this allogeneic recognition). For these experiments, freshly obtained unfractionated lymphocytes from lymph nodes and select tissues at necropsy, including spleen and liver, were washed 3 times in saline before intravenous infusion, with rhesus macaques receiving between 6 × 106 and 1 × 108 cells over 1 h (note: the number of cells transferred determined by availability, up to 1 × 108 maximum). Recipient rhesus macaques are followed for a minimum of 130 d for the onset of plasma viremia, although to date the onset of SIV viremia, if it occurs, has always ensued in the first 2 weeks post-transfer.
Vectors and viruses
The construction and characterization of the strain 68.1-derived RhCMV/SIV vectors, including RhCMV(Gag), RhCMV(retanef), RhCMV(Env), RhCMV(Pol-1), RhCMV(Pol-2) and RhCMV vector-expressing M.tuberculosis Ag85B-ESAT6 fusion protein (control) have been previously described14,16,17. RhCMV vector stocks were titered using primary rhesus fibroblasts in a tissue culture infectious dose 50 assay. The pathogenic SIV challenge stocks used in these experiments were generated by expanding SIVmac239X23 in rhesus macaque PBMCs, and were titered by using the CMMT-CD4-LTR-β-Gal (sMAGI) cell assay (NIH AIDS Reagent Program).
Viral detection assays
Plasma SIV RNA levels were determined using a gag-targeted quantitative real-time/digital RT–PCR format assay, essentially as previously described, with 6 replicate reactions analyzed per extracted sample for assay thresholds of 30 or 15 SIV RNA copies ml−129,41. Ultrasensitive determinations of plasma SIV RNA were measured by concentrating virus from larger volumes of plasma by centrifugation. For ultrasensitive measurements, typically, 1.7 ml plasma was centrifuged in a refrigerated microfuge (21,000 g, 1 h, 4 °C), nucleic acid extracted from pellets as described42, and qPCR with reverse transcription performed, with 12 reactions per extracted sample. Samples that did not yield any positive results across the replicate reactions were reported as a value of ‘less than’ the value that would apply for 1 positive reaction out of 1229. As performed, the ultrasensitive assay provided a threshold sensitivity of 1 copy ml−1 plasma for a 1.7 ml sample. Quantitative assessment of SIV DNA and RNA in cells and tissues was performed using gag-targeted, nested quantitative hybrid real-time/digital RT–PCR and PCR assays, as previously described15,29. SIV RNA or DNA copy numbers were normalized based on quantitation of a single copy rhesus genomic DNA sequence from the CCR5 locus from the same specimen, as described43, to allow normalization of SIV RNA or DNA copy numbers per 106 diploid genome cell equivalents. Ten replicate reactions were performed with aliquots of extracted DNA or RNA from each sample, with two additional spiked internal control reactions performed with each sample to assess potential reaction inhibition. Samples that did not yield any positive results across the replicate reactions were reported as a value of ‘less than’ the value that would apply for 1 positive reaction out of 10. Threshold sensitivities for individual specimens varied as a function of the number of cells or amount of tissue available and analyzed; for graphing consistency values are plotted with a common nominal sensitivity threshold. The presence of replication-competent SIV in mononuclear cell preparations derived from different tissue sites was detected by coculture of 2.5 × 105 unfractionated cells from each tissue with 1 × 105 CEMx174 cells. CEMx174 cells were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH and periodically screened for mycoplasma contamination using the Lonza MycoAlert Detection Kit, LTZ07-218. While formal authentication of this cell line was not performed in our laboratory, CEMx174 cells were used at low passage number and regularly monitored for their ability to support SIV infection. In addition, the presence of inducible replication-competent SIV was detected by coculture of 5 × 105 sort-purified CD4+ T cells isolated from lymph nodes or spleen with 1 × 105 CEMx174 cells after 12-hour stimulation with PMA (50 ng ml−1) plus ionomycin (1 μg ml−1). After 17–21 d, each culture was stained with anti-CD3 (SP34-2: Pacific Blue; BD Biosciences, 624034), anti-CD4 (L200: PerCP-Cy5.5; BD Biosciences, 624062), and intracellular SIVgag-p27 (clone 55-2F12, NIH AIDS Research and Reference Reagent Program; 55-2F12 was conjugated with FluoReporter FITC Protein Labeling Kit (Life Technologies) and used at 100 ng test−1), with positive cultures based on ≥0.5% CEMx174 cells with intracellular SIV Gag expression over background by flow cytometry3,32.
Immunological assays
SIV-specific CD4+ and CD8+ T cell responses were measured in blood and tissues by flow cytometric intracellular cytokine analysis, as previously described15,29. Briefly, mixes of sequential (11 amino acid overlapping) 15-mer peptides (AnaSpec) spanning the SIVmac239 Gag, Env, Pol, Nef, Rev, Tat, Vif, Vpr, and Vpx proteins or individual SIV Gag supertope peptides (Gag53211-222 (AADWDLQHPQP), Gag69276-284 (RMYNPTNIL), Gag73290-301 (PKEPFQSYVDRF), Gag120482-490 (EKQRESREK)) were used as antigens in conjunction with anti-CD28 (CD28.2, Purified 500 ng test−1: eBioscience, Custom Bulk 7014-0289-M050) and anti-CD49d stimulatory monoclonal antibody (9F10, Purified, 500 ng test−1: eBioscience, Custom Bulk 7014-0499-M050). Cells were incubated at 37 °C with peptide mixes and antibodies for 1 h, followed by an additional 8 h incubation in the presence of Brefeldin A (5 μg ml−1; Sigma-Aldrich). Stimulation in the absence of peptides served as background control. After incubation, stimulated cells were stored at 4 °C until staining with combinations of fluorochrome-conjugated monoclonal antibodies, including: anti-CD3 (SP34-2: Pacific Blue; BD Biosciences, Custom Bulk 624034 and PerCP-Cy5.5; BD Biosciences, Custom Bulk 624060), anti-CD4 (L200: FITC; BD Biosciences, Custom Bulk 624044 and AmCyan; BD Biosciences, Custom Bulk 658025), anti-CD8α (SK1: APC-Cy7; eBioscience, Custom Bulk 7047-0087-M002), anti-TNF-α (MAB11: APC; BD Biosciences, Custom Bulk 624076 and FITC; BD Biosciences, Custom Bulk 624046 and PE; BD Biosciences, Custom Bulk 624049), anti- IFN-γ (B27: APC; BD Biosciences, Custom Bulk 624078 and FITC; BD Biosciences, 554700), and anti-CD69 (FN50: PE; eBioscience, Custom Bulk CUST01282 and PE-TexasRed; BD Biosciences, Custom Bulk 624005). Data were collected on an LSR-II flow cytometer (BD Biosciences). Analysis was performed using FlowJo software (Tree Star). In all analyses, gating on the lymphocyte population was followed by the separation of the CD3+ T cell subset and progressive gating on CD4+ and CD8+ T cell subsets. Antigen-responding cells in both CD4+ and CD8+ T cell populations were determined by their intracellular expression of CD69 and either or both of the cytokines IFN-γ and TNF. After subtracting background, the raw response frequencies were memory corrected, as previously described14,16. To determine the phenotype of T cells and monocytes, whole blood was stained as previously described22 using these antibodies: anti-CD3 (SP34-2: Alexa700; BD Biosciences, Custom Bulk 624042 and Pacific Blue; BD Biosciences, Custom Bulk 624034), anti-CD4 (L200: AmCyan; BD Biosciences, Custom Bulk 658025), anti-CD8α (DK25: Pac Blue; DAKO, PB98401-1), anti-CD8α (SK1: APC-Cy7; eBioscience, Custom Bulk 7047-0087-M002), anti-CD95 (DX2: PE; Life Technologies, Custom Bulk CUST00525), anti-CD28 (CD28.2: PE-TexRed; BD Biosciences, Custom Bulk 624005), anti-CCR5 (3A9: APC; BD Biosciences, Custom Bulk 624046), anti-Ki67 (B56: FITC; BD Biosciences, Custom Bulk 624046), anti-CD14 (M5E2: FITC; BD Biosciences, Custom Bulk 624046), anti-CD169 (7-239: PE; BioLegend, Custom Bulk 346004), anti-CD123 (7G3: PerCP-Cy5.5; BD Biosciences, Custom Bulk 624060), anti-CD16 (3G8: Pac Blue; BD Biosciences, Custom Bulk 624034), anti-CD11c (3.9: APC; Life Technologies, Custom Bulk 7017-0116-M010), anti-HLA-DR (L243: PE-TexRed; BD Biosciences, Custom Bulk 624004), anti-CD20 (L27: APC-Cy7; BD Biosciences, Custom Bulk 655118), anti-CCR7 (15053: Biotin; R&D Systems, MAB197), and anti-streptavidin (Pac Blue; Life Technologies, S11222). Monocyte activation was determined by expression of CD169 on CD14+ HLA-DR+ CD20− CD3− CD16− cells (see Supplementary Fig. 2).
Sample size and treatment assignment
Sample size was determined by logistical and resource considerations. Treatment assignments (RhCMV/SIV versus control vaccination) were conducted after 84 dpi by rank-ordering pvl AUC (0–84 dpi) and assigning alternating treatments arbitrarily and without revision. No animals were excluded from any analysis in this study. No blinding was possible due to the constraints of working with rhesus macaques.
Statistics
All statistical analyses were conducted in R version 3.3.3 using the following R package versions: coin 1.1-3, data.table 1.10.4, ggplot2 2.2.1, gridExtra 2.2.1, knitr 1.17, lattice 0.20-34, plyr 1.8.4, pracma 2.0.7, reshape2 1.4.2, Rmisc 1.5, scales 0.4.1, stringr 1.2.0, and survival 2.40-1. Non-parametric tests were used for all statistical analyses except for the comparison of two binomial proportions (% + SIV cocultures; Fig. 5), which was conducted by chi-squared test with Yates continuity correction. The non-parametric testing included Wilcoxon tests for (paired and unpaired) comparisons and Kruskal–Wallis for more than two groups. All correlations were computed using Spearman rank correlations with significance computed using approximate distribution in presence of ties. In presence of censored data, correlations and their significance were calculated under the most conservative (least correlated) possible configuration of censored failure times. Log-rank tests were used to compare time-to-event data. All reported P values are based on two-sided tests and unadjusted except where noted. Adjusted P values were computed using the Holm procedure for family-wise error rate control. Intracellular cytokine staining data were log10-transformed before analysis, before computing AUC. For log transformations when zeros were present, a small positive constant smaller than any non-zero value was added to all values before log transformation. pvls (SIV RNA and DNA) and neutralizing antibody titers were also log10-transformed because they appeared less skewed and/or closer to the bell-shaped curve after such transformation. All transformations were conducted before computation of AUC.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The computer code used to perform statistical analysis is available from the corresponding authors on reasonable request.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Churchill, M. J. et al. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 14, 55–60 (2016).
Del Prete, G. Q. & Lifson, J. D. Considerations in the development of nonhuman primate models of combination antiretroviral therapy for studies of AIDS virus suppression, residual virus, and curative strategies. Curr. Opin. HIV AIDS 8, 262–272 (2013).
Fukazawa, Y. et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat. Med. 21, 132–139 (2015).
Miles, B. & Connick, E. TFH in HIV latency and as sources of replication-competent virus. Trends Microbiol. 24, 338–344 (2016).
Murray, A. J. et al. The latent reservoir for HIV-1: How immunologic memory and clonal expansion contribute to HIV-1 persistence. J. Immunol. 197, 407–417 (2016).
Announcement. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection-drug use, or other nonoccupational exposure to HIV—United States, 2016. MMWR Morb. Mortal. Wkly Rep. 65, 458 (2016).
Lifson, J. D. et al. Containment of simian immunodeficiency virus infection: Cellular immune responses and protection from rechallenge following transient postinoculation antiretroviral treatment. J. Virol. 74, 2584–2593 (2000).
Whitney, J. B. et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 512, 74–77 (2014).
Luzuriaga, K. et al. Viremic relapse after HIV-1 remission in a perinatally infected child. N. Engl. J. Med. 372, 786–788 (2015).
Henrich, T. J. et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 14, e1002417 (2017).
Colby, D. J. et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 24, 923–926 (2018).
Luzuriaga, K. et al. Absent HIV-specific immune responses and replication-competent HIV reservoirs in perinatally infected youth treated from infancy: Towards cure. In 20th Conference on Retroviruses and Opportunistic Infections; March 3–6, Atlanta, GA, abstract 171LB (2013).
Persaud, D. et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N. Engl. J. Med. 369, 1828–1835 (2013).
Hansen, S. G. et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473, 523–527 (2011).
Hansen, S. G. et al. Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013).
Hansen, S. G. et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 15, 293–299 (2009).
Hansen, S. G. et al. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340, 1237874 (2013).
Hansen, S. G. et al. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science 351, 714–720 (2016).
Kim, W. K. et al. Increased expression of CD169 on blood monocytes and its regulation by virus and CD8 T cells in macaque models of HIV infection and AIDS. AIDS Res. Hum. Retrovir. 31, 696–706 (2015).
van der Kuyl, A. C. et al. Sialoadhesin (CD169) expression in CD14+ cells is upregulated early after HIV-1 infection and increases during disease progression. PLoS One 2, e257 (2007).
Nowak, M. A. et al. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71, 7518–7525 (1997).
Del Prete, G. Q. et al. Short communication: Comparative evaluation of coformulated injectable combination antiretroviral therapy regimens in simian immunodeficiency virus-infected rhesus macaques. AIDS Res. Hum. Retrovir. 32, 163–168 (2016).
Del Prete, G. Q. et al. Molecularly tagged simian immunodeficiency virus SIVmac239 synthetic swarm for tracking independent infection events. J. Virol. 88, 8077–8090 (2014).
Okoye, A. et al. Profound CD4+/CCR5+ T cell expansion is induced by CD8+ lymphocyte depletion but does not account for accelerated SIV pathogenesis. J. Exp. Med. 206, 1575–1588 (2009).
Schmitz, J. E. et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283, 857–860 (1999).
Barouch, D. H. et al. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science 349, 320–324 (2015).
Liu, J. et al. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science 353, 1045–1049 (2016).
Henrich, T. J. et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 207, 1694–1702 (2013).
Hansen, S. G. et al. Addendum: Immune clearance of highly pathogenic SIV infection. Nature 547, 123–124 (2017).
Bruner, K. M. et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 22, 1043–1049 (2016).
Ho, Y. C. et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551 (2013).
Fukazawa, Y. et al. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat. Med. 18, 1673–1681 (2012).
Bui, J. K. et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Pathog. 13, e1006283 (2017).
Hosmane, N. N. et al. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 214, 959–972 (2017).
Kim, M. & Siliciano, R. F. Reservoir expansion by T-cell proliferation may be another barrier to curing HIV infection. Proc. Natl Acad. Sci. USA 113, 1692–1694 (2016).
Kwon, K. J. & Siliciano, R. F. HIV persistence: Clonal expansion of cells in the latent reservoir. J. Clin. Invest. 127, 2536–2538 (2017).
Maldarelli, F. et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183 (2014).
Simonetti, F. R. et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl Acad. Sci. USA 113, 1883–1888 (2016).
Hessell, A. J. et al. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat. Med. 22, 362–368 (2016).
Loffredo, J. T. et al. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81, 8827–8832 (2007).
Li, H. et al. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl Acad. Sci. USA 113, E3413–E3422 (2016).
Cline, A. N. et al. Highly sensitive SIV plasma viral load assay: Practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34, 303–312 (2005).
Venneti, S. et al. Longitudinal in vivo positron emission tomography imaging of infected and activated brain macrophages in a macaque model of human immunodeficiency virus encephalitis correlates with central and peripheral markers of encephalitis and areas of synaptic degeneration. Am. J. Pathol. 172, 1603–1616 (2008).
Acknowledgements
This work was supported by the US National Institutes of Health (grants U19AI096109, U19AI095985, UM1AI126611, UM1AI124377, R37AI054292, and P51OD011092, L.J.P.), by the Bill and Melinda Gates Foundation (grant OPP1094567, L.J.P.), and supported in part with federal funds from the National Cancer Institute, National Institutes of Health (Contract No. HHSN261200800001E, J.D.L.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. The authors thank B. Keele (Leidos Biomedical Research, Inc.) for providing SIVmac239X and Janssen Pharmaceuticals for providing Darunavir. The CD8+ lymphocyte-depleting monoclonal antibody, M-T807R1, was provided by the National Institutes of Health’s Nonhuman Primate Reagent Resource Program. We thank A. Sylwester, S. Hagen, T. Swanson, M. Fischer, S. Planer, C. Kahl, D. Siess, M. Reyes, J. Clock, A. Konfe, C. Abana, C. Pexton, E. McDonald, K. Jeffries, M. Grey, C. Xu, W. Brantley, A. Maxwell, M. Lidell, D. Malouli, M. Marenco, A. Townsend, and L. Boshears for technical or administrative assistance.
Author information
Authors and Affiliations
Contributions
L.J.P. and J.D.L. conceived of the study and wrote the paper with assistance from A.A.O. A.A.O. and S.G.H. managed the project, performed all animal experiments, and analyzed immunological and virological data, assisted by M.V., Y.F., H.P., D.M.D., R.L., C.M.H., A.B.V., E.A., J.C.F., D.M., and R.M.G. J.D.L. planned and performed SIV quantification assisted by K.O., R.S., R.F., and W.J.B. A.W.L. and M.K.A. managed the animal protocols and J.D.L., J.H., and R.G. developed the injectable cART formulation. P.T.E. and B.R.B. conducted all statistical analyses and contributed to the writing of the paper.
Corresponding authors
Ethics declarations
Competing interests
Oregon Health & Science University, L.J.P., and S.G.H. have a financial interest in Vir Biotechnology, Inc., a company that may have a commercial interest in the results of this research and technology. The potential individual and institutional conflicts of interest have been reviewed and managed by Oregon Health & Science University.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Table 1
Rights and permissions
About this article

Cite this article
Okoye, A.A., Hansen, S.G., Vaidya, M. et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat Med 24, 1430–1440 (2018). https://doi.org/10.1038/s41591-018-0130-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-018-0130-7
This article is cited by
-
Exploring synergies between B- and T-cell vaccine approaches to optimize immune responses against HIV—workshop report
npj Vaccines (2024)
-
More than the Infinite Monkey Theorem: NHP Models in the Development of a Pediatric HIV Cure
Current HIV/AIDS Reports (2024)
-
Early antiretroviral therapy favors post-treatment SIV control associated with the expansion of enhanced memory CD8+ T-cells
Nature Communications (2024)
-
Prevention, treatment and cure of HIV infection
Nature Reviews Microbiology (2023)
-
Advances in HIV therapeutics and cure strategies: findings obtained through non-human primate studies
Journal of NeuroVirology (2023)
