
Abstract
Small-molecule inhibitors of the serine dipeptidases DPP8 and DPP9 (DPP8/9) induce a lytic form of cell death called pyroptosis in mouse and human monocytes and macrophages1,2. In mouse myeloid cells, Dpp8/9 inhibition activates the inflammasome sensor Nlrp1b, which in turn activates pro-caspase-1 to mediate cell death3, but the mechanism of DPP8/9 inhibitor-induced pyroptosis in human myeloid cells is not yet known. Here we show that the CARD-containing protein CARD8 mediates DPP8/9 inhibitor-induced pro-caspase-1-dependent pyroptosis in human myeloid cells. We further show that DPP8/9 inhibitors induce pyroptosis in the majority of human acute myeloid leukemia (AML) cell lines and primary AML samples, but not in cells from many other lineages, and that these inhibitors inhibit human AML progression in mouse models. Overall, this work identifies an activator of CARD8 in human cells and indicates that its activation by small-molecule DPP8/9 inhibitors represents a new potential therapeutic strategy for AML.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
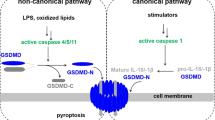
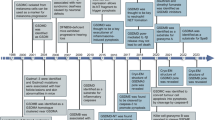

References
Okondo, M. C. et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol. 13, 46–53 (2017).
Taabazuing, C. Y., Okondo, M. C. & Bachovchin, D. A. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem. Biol. 24, 507–514.e4 (2017).
Okondo, M. C. et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem. Biol. 25, 262-267.e5 (2018).
Zhang, H., Chen, Y., Keane, F. M. & Gorrell, M. D. Advances in understanding the expression and function of dipeptidyl peptidase 8 and 9. Mol. Cancer Res. 11, 1487–1496 (2013).
Rosenblum, J. S. & Kozarich, J. W. Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr. Opin. Chem. Biol. 7, 496–504 (2003).
Wagner, L., Klemann, C., Stephan, M. & von Horsten, S. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin. Exp. Immunol. 184, 265–283 (2016).
Adams, S. et al. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 64, 5471–5480 (2004).
Walsh, M. P. et al. Val-boroPro accelerates T cell priming via modulation of dendritic cell trafficking resulting in complete regression of established murine tumors. PLoS One 8, e58860 (2013).
Cunningham, C. C. Talabostat. Expert Opin. Investig. Drugs 16, 1459–1465 (2007).
Eager, R. M. et al. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin. Oncol. 21, 464–472 (2009).
Eager, R. M. et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer 9, 263 (2009).
Lankas, G. R. et al. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes 54, 2988–2994 (2005).
Jiaang, W. T. et al. Novel isoindoline compounds for potent and selective inhibition of prolyl dipeptidase DPP8. Bioorg. Med. Chem. Lett. 15, 687–691 (2005).
Wu, J. J. et al. Biochemistry, pharmacokinetics, and toxicology of a potent and selective DPP8/9 inhibitor. Biochem. Pharmacol. 78, 203–210 (2009).
Bachovchin, D. A. et al. A high-throughput, multiplexed assay for superfamily-wide profiling of enzyme activity. Nat. Chem. Biol. 10, 656–663 (2014).
Spagnuolo, P. A. et al. Inhibition of intracellular dipeptidyl peptidases 8 and 9 enhances parthenolide’s anti-leukemic activity. Leukemia 27, 1236–1244 (2013).
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012).
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E. & Monack, D. M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Guey, B., Bodnar, M., Manie, S. N., Tardivel, A. & Petrilli, V. Caspase-1 autoproteolysis is differentially required for NLRP1b and NLRP3 inflammasome function. Proc. Natl Acad. Sci. USA 111, 17254–17259 (2014).
Van Opdenbosch, N. et al. Activation of the NLRP1b inflammasome independently of ASC-mediated caspase-1 autoproteolysis and speck formation. Nat. Commun. 5, 3209 (2014).
Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309 (2011).
Bagger, F. O. et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 44, D917–924 (2016).
Ting, J. P. et al. The NLR gene family: a standard nomenclature. Immunity 28, 285–287 (2008).
D’Osualdo, A. et al. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One 6, e27396 (2011).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Razmara, M. et al. CARD-8 protein, a new CARD family member that regulates caspase-1 activation and apoptosis. J. Biol. Chem. 277, 13952–13958 (2002).
Frew, B. C., Joag, V. R. & Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659 (2012).
Van Goethem, S. et al. Inhibitors of dipeptidyl peptidase 8 and dipeptidyl peptidase 9. Part 2: isoindoline containing inhibitors. Bioorg. Med. Chem. Lett. 18, 4159–4162 (2008).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Khwaja, A. et al. Acute myeloid leukaemia. Nat. Rev. Dis. Primers 2, 16010 (2016).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014).
Coutts, S. J. et al. Structure–activity relationships of boronic acid inhibitors of dipeptidyl peptidase IV. 1. Variation of the P2 position of Xaa-boroPro dipeptides. J. Med. Chem. 39, 2087–2094 (1996).
Wang, K. et al. Patient-derived xenotransplants can recapitulate the genetic driver landscape of acute leukemias. Leukemia 31, 151-158 (2016).
Daigle, S. R. et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 (2011).
Acknowledgements
We thank S. Monette for histology advice, and W. Bachovchin, W. Wu and J. Lai (Tufts University, USA) for Val-boroPro, 1G244, 8j and l-allo-Ile-isoindoline. This work was supported by the Josie Robertson Foundation (D.A.B. and A.K.), a Stand Up to Cancer-Innovative Research Grant (grant number SU2C-AACR-IRG11-17 to D.A.B.; Stand Up to Cancer is a program of the Entertainment Industry Foundation; research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C), the Pew Charitable Trusts (D.A.B. is a Pew-Stewart Scholar in Cancer Research), the NIH (R01 AI137168 to D.A.B.; R21 CA188881 and R01 CA204396 to A.K.; T32 GM115327-Tan to D.C.J.; the MSKCC Core Grant P30 CA008748), the American Cancer Society (Postdoctoral Fellowship PF-17-224-01 – CCG to C.Y.T.), Borroughs Wellcome Fund (A.K.), Alex’s Lemonade Stand Foundation (A.K.), Gabrielle’s Angel Foundation (A.K.) and the Damon Runyon Cancer Research Foundation (A.K. is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator).
Author information
Authors and Affiliations
Contributions
D.A.B. conceived and directed the project, performed experiments, analyzed data and wrote the paper; D.C.J., C.Y.T., M.C.O., A.J.C. and S.D.R. performed experiments and analyzed data; D.C.J., M.C.O., F.C.B., C.R. and E.P. performed xenograft experiments; E.d.S. advised on the xenograft experiments. A.K. provided cell biology, mouse xenograft and leukemia expertise.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–16 and Supplementary Tables 1–4
Rights and permissions
About this article

Cite this article
Johnson, D.C., Taabazuing, C.Y., Okondo, M.C. et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat Med 24, 1151–1156 (2018). https://doi.org/10.1038/s41591-018-0082-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-018-0082-y
This article is cited by
-
Crosstalk between metabolism and cell death in tumorigenesis
Molecular Cancer (2024)
-
Mechanistic insights from inflammasome structures
Nature Reviews Immunology (2024)
-
New prospects of cancer therapy based on pyroptosis and pyroptosis inducers
Apoptosis (2024)
-
Involvement of inflammasomes in tumor microenvironment and tumor therapies
Journal of Hematology & Oncology (2023)
-
NLRP1- A CINDERELLA STORY: a perspective of recent advances in NLRP1 and the questions they raise
Communications Biology (2023)
