
Abstract
The immune system is highly complex and distributed throughout an organism, with hundreds to thousands of cell states existing in parallel with diverse molecular pathways interacting in a highly dynamic and coordinated fashion. Although the characterization of individual genes and molecules is of the utmost importance for understanding immune-system function, high-throughput, high-resolution omics technologies combined with sophisticated computational modeling and machine-learning approaches are creating opportunities to complement standard immunological methods with new insights into immune-system dynamics. Like systems immunology itself, immunology researchers must take advantage of these technologies and form their own diverse networks, connecting with researchers from other disciplines. This Review is an introduction and ‘how-to guide’ for immunologists with no particular experience in the field of omics but with the intention to learn about and apply these systems-level approaches, and for immunologists who want to make the most of interdisciplinary networks.
Similar content being viewed by others


Main
Systems immunology provides a holistic understanding of the immune system, spanning single immunological components and pathways to form cross-scale networks. Unlike reductionist approaches aimed at understanding individual parts, systems immunology aims to understand properties of individual parts working together—a challenge requiring specialized methodologies1,2.
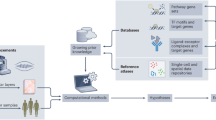
During the past century, many successful experimental strategies have been developed that were instrumental in defining cell types and cellular states within the immune system to reveal major molecular and functional components of the immune system and to establish causal relationships for transcriptional and functional cascades that drive immune activation (Fig. 1). For two decades, high-throughput, high-resolution technologies from the omics field have revolutionized our understanding of immunology and enabled the simultaneous assessment of hundreds to thousands of cellular, functional and molecular parameters with continuously increasing throughput and decreasing turn-around times.

Timeline of the most important technological developments in immunology research, with a special focus on the evolution of omics from the advent of microarrays to current single-cell approaches.
Sequencing-based technologies are used to assess genomic, transcriptomic and epigenomic information, and sophisticated technologies in proteomics, metabolomics, microbiomics and lipidomics have been introduced to immunological research. In the past decade, single-cell sequencing technologies have emerged, with single-cell transcriptomics leading the way3.
In this how-to guide, we provide a brief introduction in the use and integration of omics technologies in systems immunology and explain how current single-cell-level omics technologies can be applied to immunological questions in model systems and increasingly in human immunology and clinical trials for immune-mediated diseases. We focus on the use of transcriptomic technologies in systems immunology, particularly on single-cell assays, as mRNA constitutes the first functional and relatively easily accessible readout of the genome; as such, it can serve as a surrogate to bridge genomic and functional phenotypes to enable the description and prediction of causal relationships and effectors of immune-cell function.
Evolution of omics in immunology
High-throughput, high-resolution omics technologies have been used in immunology since microarrays were introduced4,5. These early microarray-based techniques were applied widely, for example to understand genetic differences and evolution of Bacillus Calmette–Guérin (BCG) vaccines6, to examine systemic inflammation and the network of leukocytes in people with systemic lupus erythematosus5 and to characterize the activation network of macrophages in response to diverse stimuli7. However, in the late 2000s, microarray-based techniques were superseded by unbiased massive-scale whole RNA sequencing (RNA-seq)8,9. The identification of long non-coding RNAs as broad-acting regulatory components of inflammatory responses exemplified how these technologies can lead to the discovery of completely new molecular concepts in immunology10. Shortly thereafter, the first single-cell immune-cell transcriptomes were described11,12, which fundamentally changed the way immune-cell types and cellular states can be defined and how this information can be used to predict cellular activity and immune-cell function. Since then, a single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq), single-cell DNA methylation, single-cell lipidomics and single-cell metabolomics have been introduced as further means to characterize immune cells13,14,15.
Specific to immunological research, repertoire analyses of recombined B cell receptors (BCRs) and T cell receptors (TCRs) by BCR-seq and TCR-seq have a crucial role in understanding the complex mechanisms controlling the diversity and specificity of adaptive immune responses16. Combined with single-cell transcriptomic and antigen-binding analyses using sophisticated analytical tools17,18,19,20, BCR-seq and TCR-seq can shed light on the functional state of the adaptive immune repertoire and its specificity in response, for example to pathogens or tumor antigens, which might become important features for diagnosis and therapy of immune-mediated diseases21.
In parallel to the advances in single-cell genomics, the field of multiparameter antibody-based characterization of immune cells has deepened our understanding of immunology, mainly owing to the advent of heavy-metals-based cytometry by time of flight (CyTOF) and oligonucleotide-based cellular indexing of transcriptomes and epitopes (CITE)-seq, complementing the development of high-dimensional fluorochrome-based flow cytometry. Furthermore, imaging mass cytometry is a highly valuable extension of CyTOF for characterizing immune cells in their natural environment and spatial context.
The first antibody-independent mass-spectrometry-based single-cell proteomics technologies, such as ScoPE2, were also reported this year and present a logical next step for the characterization of immune cells22.
As technologies continue to improve, metabolome, proteome, microbiome and lipidome studies and the integration of the data that they produce are being used to tackle immunological questions at a system-wide scale. Nevertheless, sequencing-based technologies remain the most commonly used techniques, particularly at the single-cell level. The most accessible omics technologies for immunology researchers are listed in Tables 1–3 and have been reviewed elsewhere23,24,25,26.
Choosing an omics technology
When it comes to the application of omics technologies in systems immunology, one needs first to define which of the omics technologies are best suited to answer the proposed question. Here, we introduce the respective technologies for the three major layers of biological information, namely the transcriptome, epigenome and genome.
Transcriptomics
Techniques for interrogating the transcriptome should be mentioned as forerunners of the omics revolution. RNA sequencing remains the gold standard for unbiased, genome-wide assessment of gene expression on a population level, and many protocols exist for a variety of purposes27.
For questions focusing on the heterogeneity of cell populations in health and disease or cellular differentiation and developmental trajectories, single-cell transcriptomics has quickly gained popularity since its introduction28. Despite the cost and technical complexity, we encourage the use of single-cell technologies when studying heterogeneous cellular populations and molecular phenotypes (for example, in complex tissues or in response to a diversity of perturbations). If preliminary results indicate negligible cellular heterogeneity, bulk analysis is a viable option given its lower cost and thereby its potential to analyze larger sample sizes and to provide higher density transcriptomic information. Furthermore, for large to very large clinical studies exceeding hundreds of samples, bulk technologies in combination with deconvolution algorithms trained on a small set of single-cell resolved data are currently an effective way to gain important information about transcriptional regulation and function (for example, associated with a specific disease), pathological processes or therapeutic interventions, including vaccines29,30,31.
Today, several complementary scRNA-seq approaches exist, each with specific advantages and applications for answering different questions (summarized in Table 1). As examples of the two most widely used scRNA-seq methods, plate-based full-length-mRNA techniques have the highest sensitivity (albeit limited in throughput) and enable isoform detection in isolated cell populations32, and 3′- or 5′-mRNA-capture approaches using microfluidics or nanoliter-well arrays enable high throughput at the cost of decreased sensitivity33,34.
Although profiling of the immune-receptor repertoire on a population level can inform us about clonal diversity of lymphocytes in homeostasis and disease16, its adaptation to single-cell resolution (scTCR-seq and scBCR-seq) enables clonotypic phenotype analysis35. Furthermore, oligonucleotide-coupled antibodies (CITE-seq36 or commercially available reagents, such as TotalSeq and AbSeq) enable the combination of scRNA-seq with analysis of surface protein expression.
Epigenomics
In addition to the transcriptome, the epigenome can be interrogated using omics technologies. The variety within this class of technologies reflects the complexity of epigenetic regulation37. Although ATAC-seq is seemingly the most prominent representative of epigenomics technologies38, many solutions to investigate DNA accessibility and conformation, histone modifications and transcription factor binding at the bulk and single-cell level have been developed and are summarized in Table 2. Furthermore, multi-omics methods to profile epigenetic markers alongside the transcriptome in single cells have undergone a rapid development from proof-of-concept reports to robust protocols applicable at a large scale39.
Genomics
Omics technologies are crucial to define genetic alterations as major drivers for human immune phenotypes (Table 3)40. Aside from whole-genome or whole-exome sequencing and targeted next-generation sequencing (NGS) panels, genotyping microarrays present a scalable and cost-effective solution for population genetics. Genetic variability has been investigated at the single-cell level (reviewed elsewhere41), but often these data suffer from sparsity and high levels of noise as the biological material assessed in a single cell is limited. Exploiting structural genomic rearrangements leading to abnormal copy numbers by copy-number variation (CNV) analyses or taking advantage of the higher copy number of the mitochondrial genome by targeted mitochondrial DNA sequencing (scMito-seq) are sophisticated approaches to overcome these limitations and can be used to infer cell fate through lineage tracing42,43,44,45.
The need for hypothesis-driven research
The potential of omics technologies in immunology seems endless, and the high-dimensional output of these technologies often triggers unbiased analytical approaches. Although exploratory data analysis can be essential to initially understand data structure and detect potential biases, we encourage researchers to follow established principles of hypothesis-driven science as outlined in the proposed systems-immunology cycle (Fig. 2)46. In this cycle, the application of (single-cell) multi-omics technologies follows the formulation of the hypothesis or question in combination with classical experimental design (for example, loss-of-function or gain-of-function experiments, defined clinical cohorts or clinical trials, such as vaccine trials or immunotherapy) to establish immune function, molecular phenotypes or immunotherapy and outcome prediction. Although a hypothesis is seen by some as a liability, we stress its guiding function while acknowledging the risk of it blinding researchers to alternative questions or paths of analysis47. Admittedly, a hypothesis in omics-based immunological studies can be vague, such as proposing broad transcriptional differences in multiple peripheral immune cells in a case–control study of an inflammatory disease. Nevertheless, we argue that a hypothesis-driven modus operandi helps scientists formulate and focus on central questions and does not preclude the potential for independent discovery and the derivation of new hypotheses in secondary data usage.

The systems-immunology cycle, with representative examples for each step, from the first medical observation or phenotype to validation of results. DE, differential expression; ML, machine learning.
The major difference between this holistic approach and classical reductionist experimentation is the requirement for mathematical and computational modeling of big data. This step of the cycle could be termed ‘data driven’; however, without a well-formulated hypothesis and sound experimental design, cutting-edge multi-omics technologies are at risk of missing their mark. By contrast, hypothesis-driven approaches and well-conceived experimental setups result in high-resolution omics data that provide valuable, and often unanticipated, biological explanations while reducing the risk of failure and enabling the prioritization of follow-up and validation studies.
How to apply omics technology
Human immunology has already benefited substantially from omics technologies, such as the large endeavors of the Human Immunology Project Consortium48, the ImmVar study49, the Human Functional Genomics Project40,50 and the Milieu Intérieur study51, to study the variability of the human immune system and to better characterize genotype-phenotype associations. Bulk omics technologies, such as DNA-seq, RNA-seq and ATAC-seq, are now commonly included in clinical studies of immunological diseases52,53. The Human Cell Atlas was the first large initiative to integrate omics technologies54, and now single-cell-resolution technologies (in particular scRNA-seq) are increasingly included in systems-level immunological readouts in large clinical trials55.
Experimental design
For human immunology research, five scenarios can be envisioned (Fig. 2): (1) exploratory studies to define immune functions and immune-cell types, usually performed in small cohorts up to 20 individuals56; (2) validation studies in humans assessing immunological findings derived from model systems43; (3) cross-sectional cohort studies, either in healthy or diseased individuals, to study human immune variation and immune deviation in the context of diseases57; (4) vaccine, immunotherapy and other therapy-response trials31; and (5) studies exploring genetic or environmental effects on human immune function58,59. Depending on the goals and the size of the study, factors affecting data quality might differ. For example, although human variation can have a strong effect on exploratory studies with samples from only a few individuals, restricted diversity within a cohort might not fully represent the spectrum of human variation for genome-wide assessments, which also holds true for genetic susceptibility studies. Similar considerations need to be included in the design of (immuno)therapy trials. For example, the assessment of the dynamics of immune activation, function and cellular distribution following a vaccine will differ between individuals, and genome-wide changes might have different kinetics, the capture of which requires not only highly standardized sampling schemes, but also sophisticated analytical approaches60,61,62.
Necessity of teamwork and expertise
Compared with the lower-resolution methods that are often used as primary readouts in clinical studies63, the application of omics technologies requires consideration of many potential factors that affect data quality, and thus requires thorough planning to harness the potential of high-resolution, high-content technologies. As technologies are continuously evolving, a team of omics experts should be included in the design of clinical studies that address immunological questions. Furthermore, omics data generation and analysis should be included in educational programs in immunology46.
In addition to study design, sample handling according to well-defined standard operating procedures (SOPs), library production, sample multiplexing, sequencing strategies and depth, data pre-processing and downstream analyses, including metadata handling, need to be addressed (Box 1).
Batch effects
One, if not the major, aspect when planning omics applications, particularly with increasing sample sizes generated across different institutions, is the consideration of the effect from technical parameters, often referred to as ‘batch’ effects64,65. Given the vast number of measurements defining the feature space, ranging from a few (~100) features in targeted sequencing up to hundreds of thousands of features in chromatin-landscape analysis, and considering the sensitivity of omics technologies, batch effects can be introduced at any step in the sample and data-generation process; thus, attempts to decrease batch effects are essential for good study design.
For single-cell transcriptomics, multiplexing of samples enables joint processing, which can help to reduce technical variability. A number of strategies for this purpose have been developed, including labeling of cells using either oligonucleotide-tagged antibodies (cell-hashing) or lipid-modified and cholesterol-modified oligonucleotides66, or the use of natural genetic variation67 to disentangle cells from multiple donors.
To avoid effects of circadian rhythm and seasonality, samples should be collected at similar times of day if possible68,69. For studies conducted over an extended period of time, seasonality might either be considered as a covariate of immune function70 or eliminated by sampling during the same time of the year. Although seemingly trivial, sampling itself needs to be highly standardized, as organ, location of biopsy, sampling devices, time from biopsy to sample processing and sample-freezing procedures need to be as uniform as possible71,72,73, and any deviations from the protocol must be recorded carefully for each included sample. Such technical and clinical metadata can later be useful to understand unanticipated variance and tackle batch effects during data analysis using batch-effect-removal algorithms65,74. In addition, if available, these metadata facilitate further reuse of the data.
Isolation procedures for RNA or DNA also require careful standardization. For example, batch effects might be introduced by handling some samples manually and others using automated sample handling. Similarly, if studies become too large to handle all samples in one run, individual batches might have differences due to the reagents or buffers that are used. Here, randomization of the samples extracted and processed in each batch can prevent the introduction of uncontrollable biases in the data.
Prior to the setup of larger (multi-center) studies, small pilot trials evaluating all necessary steps and predicting potential confounding effects of upscaling are advisable.
Taken together, batch effects have to be considered in the interpretation of omics data, and knowing their origin and how to minimize their effect on data analysis is critical for the production of robust results.
Metadata collection and standardized documentation
Collecting dense technical and clinical metadata on participants in clinical trials when using omics technologies is becoming more important. Interpretability of variability observed in high-resolution omics data might remain opaque without comprehensive records covering both technical aspects, such as sampling method and device, library production protocol or experimental day or batch, and clinical parameters, including sex, age, body-mass index, disease history, comorbidities, medication, smoking history and additional clinical markers such as serum levels of inflammatory biomarkers and differential blood-cell counts (see refs. 57,75,76). Even worse, missing technical or clinical metadata might cause misinterpretation of complex data and mislead subsequent research directions. Also crucial for meta-analysis is that researchers and publishers enable metadata to be accessible while respecting privacy and data-protection regulations. Moreover, the use of accepted ontologies for clinical metadata helps to maintain the highest possible degree of consistency across studies77.
In principle, similar caution should be applied when it comes to sequencing, as library production, and even sequencing itself, have many variables that affect downstream analysis (Box 1). Although sequencing core facilities usually know how to minimize batch effects, careful planning of these steps together can improve data quality.
Aside from the many pitfalls in data production, data processing and analysis also require high standards for reproducibility and documentation. The many options and consequential choices during data processing have noticeable effects on data content and quality and therefore need to be standardized for any given project and stringently reported to the community. Taking the simple example of the alignment of sequencing reads to a reference genome, it has been thoroughly demonstrated how the choice of the alignment algorithm and reference genome and transcriptome annotation can affect data quality and content78. Another clear but important example is the selection of gene biotypes, such as protein-coding, long non-coding or microRNAs, in gene expression quantification and downstream analyses. Focusing on protein-coding genes, as is quite common in transcriptomic analyses, might simplify the task of analyzing and interpreting gene expression data, but prevents assessment of regulation mediated by non-coding RNAs, despite the fact that total RNA libraries contain this information.
Sample size
Sample size is another important aspect to be considered in the design of omics studies in human-systems immunology. Although exploratory pilot studies work with low numbers of well-defined samples, studies addressing genetic susceptibility require large cohorts. Furthermore, the contrast of the inter-individual variability versus the intra-individual (that is, inter-cellular) heterogeneity needs to be specifically considered for sample-size estimation in single-cell omics studies. Molecular profiling of a specific subset of cells from individuals with a heterogeneous disease requires many samples from a relatively large patient cohort. By comparison, an exploratory study of a clinically well-defined disease spanning a whole cellular compartment, such as peripheral blood immune cells and their cell states, will require large numbers of cells from each individual.
Moreover, the fact that the effect sizes among different features can vary considerably further complicates study size estimation. Approaches to power calculation, such as the scPower or powsimR frameworks79,80, should be taken into account during study design.
The high cost of sequencing-based omics techniques still limits the number of samples possible to analyze. One potential solution to avoid underpowered studies is to focus on subcohorts of individuals in which the largest effect size is predicted to evaluate the initial hypothesis considering intra-sample and inter-sample heterogeneity and to include additional individuals only if an interim analysis has shown differences between the groups. Multiplexing samples can substantially reduce cost and is particularly suitable for studies with many samples but that require few cells per sample. In addition, the initial sequencing data are used to perform a better power estimation and, if necessary, sequencing data from additional samples have to be added to the study data set. This is particularly feasible for omics data for which samples can be safely stored for extended periods of time. This approach enables optimization between cost and the informative value of the study, as long as technical batch effects between study phases are minimized.
Challenges and opportunities for data analysis
Once data production and quality control have been completed, in-depth downstream analysis of the data can begin. In Box 2, we list the hardware requirements for such analyses and a selection of bioinformatics tools that we find particularly useful. With the vast numbers of new bioinformatics tools and the fast pace at which they are being published, the possibilities for analyses are seemingly endless81. We therefore strongly advocate for the formulation of an analysis plan with clearly defined and prioritized questions and well-established methods to address them (Fig. 3), if possible in consultation with experienced data scientists. Such a plan can greatly speed up the analysis and help computational team members who might not have the subject-specific biological knowledge to address the most relevant questions. In view of the enormous feature space and large amount of room for unexpected observations, this plan must be dynamic. But, even if it seems naive, writing a strategy that allows for adjustments prevents the analyst from getting lost in the many analytical possibilities. As already emphasized, we favor hypothesis-driven studies that combine omics data with computational modeling and experimental validation as a powerful approach for data generation, interpretation and efficient knowledge gain. Once priorities and major questions are defined, and depending on the nature of the available data—be it transcriptomic, epigenetic or genetic data at bulk or single-cell resolution—different analytical pipelines and tools need to be applied (reviewed elsewhere26,81). First analytical steps comprise means of data exploration using unbiased and unsupervised methodologies, such as hierarchical clustering or principal component analysis. Understanding the data independent of the initial hypothesis is vital to identify the present axes of variance in uncharted data and to grasp the dominating variables, such as disease classification in clinical studies or the experimental date in case of batch effects. Evaluation of the robustness of parameter settings (for example, quality filtering cut-offs, doublet identification, dimensionality-reduction parameters and clustering resolutions) and establishment of a suitable data model might take considerable time and should not be underestimated.

Proposed workflow for experimental and analytical planning in the systems-immunology cycle.
Subsequently, hypothesis testing using statistical methods to contrast gene expression levels in different groups within the study cohort presents a major readout82. An alternative to classical inferential hypothesis testing to define differentially expressed genes between groups of samples are gene regulatory network approaches that enable identification of subtle, but robust, patterns of expression changes within the study despite small effect sizes83,84. Naturally, with the introduction of single-cell resolution in omics technologies, the complexity of data analysis has increased. New challenges have emerged, including unprecedented dimensionality as well as high sparsity and noise in the data. In addition, cell-type identification and annotation in the context of existing knowledge, integration of data across experiments or cell-type-associated expression of genes has demanded new analytical solutions85.
The results of such complex computer-aided analyses can be viewed as models of the data, which depend on numerous parameter settings and aim to represent the underlying biology. This is probably the major difference compared with classical readouts (for example, a cytokine measurement or cell surface marker expression). Uncertainties are inherent to all of these computational approaches and are controlled in two complementary ways. First, the use of different computational methods to describe the data structure presents an in silico validation of the model. Second, experimental validations are crucial, for example to test predicted cellular phenotypes by functional assays in the laboratory.
However, to propose such validation experiments, the data models must be biologically interpreted. Questions to be asked range from defining signaling pathways changed within the study cohort86, predicting transcription factor binding within regions of open chromatin inducing certain transcriptional programs87, to modeling potential ligand-receptor interactions between different cell types, if single-cell data are available88,89. For almost all of these important tasks, many approaches and tools are available. When considering how to apply new predictive layers to unravel underlying biology within the data, it is often advisable to collaborate or exchange information with experts who have introduced new computational approaches.
Machine learning
Attempts to integrate multiple omics layers, for example transcriptomic with metabolomic90 or epigenomic data91, add yet another level of complexity to the analysis. As expected, the mathematical and computational models for integration are complicated and constantly evolving. We expect new, innovative and user-friendly approaches to be introduced in the next few years92. This is similarly true when it comes to machine-learning methods that are becoming more common for the analysis of omics data93, particularly at the single-cell level81. As machine learning is also a very wide and strongly proliferating field, it is of utmost importance that the question to be answered is phrased clearly before applying machine-learning strategies. Reconstruction of gene regulatory networks to identify targetable hub genes is one possible application of machine learning87. Moreover, for the characterization of transcriptional alterations induced by, for example, a new infectious disease, deep-learning strategies can be used to map new data sets on top of a reference from healthy individuals94.
Data storage
By definition, omics data sets are large, and with regard to large human cohorts or clinical studies, data storage and processing require a lot of computing resources (Box 2). Moreover, omics data contain information that can be sufficient to re-identify individuals and therefore are regulated by national laws for privacy protection, such as the General Data Protection Regulation (GDPR) in the European Union and the Health Insurance Portability and Accountability Act (HIPAA) in the United States. It is therefore advisable to take the necessary ethical and legal precaution when dealing with human omics data. Data storage needs to occur either on highly protected in-house systems or on regulated repositories maintained by public organizations, such as the European Genome Archive (EGA) or the database of Genotypes and Phenotypes (dbGaP). Pre-processing of raw data, including quality control, alignment or pseudoalignment to reference genomes and transcriptomes or novel assemblies, and normalization of data, is computationally expensive, requiring significant compute power, which is best carried out by collaborating genome centers. Once data are preprocessed and summarized, the data size is usually smaller, and such data can be handled by standard computer equipment that is even as low-powered as laptops. Irrespective of the computing infrastructure used for analysis, data-privacy standards should be recognized at all times, and platforms integrating user management, data access, data and metadata management, data storage and data analysis in a protective environment are the way forward. Such platforms can provide principles of findability, accessibility, interoperability and reusability (FAIR)95 and containerized environments to ensure data reproducibility with the option of serving as a safe place to train young scientists in computational biology applications96.
Data availability
Given the high computational component of current omic studies, the respective code and scripts are a critical component of the work. Although it is impractical to publish the entire code in the materials section of a publication, we strongly encourage the community to provide the entire source code used for the analysis in public repositories (for example GitHub or Zenodo) to ensure full reproducibility of analyses. Furthermore, online platforms such as FastGenomics96 provide a place to store both preprocessed data and the accompanying code, allowing the reader to interactively reproduce the analysis in predefined containerized environments.
When it comes to data sharing, access and reuse, the omics field is currently leading the way147 and it is good to see that similar strategies are now being supported within the field of immunology research, for example for CyTOF and multiparameter flow cytometry data or immunophenotyping data for clinical studies148. Benchmarking new studies, or using previous knowledge to classify insights from new omics data, is becoming a standard procedure. This rich information from existing data can be further leveraged. For example, cohort-wide data sets of functional immunological and omics information can be used to assess human variation in gene expression as a predictor for gene function when comparing individuals with low or high expression of a gene of interest97.
Validation of omics data
Omics data are of value during the experimental validation phase of the systems-immunology cycle (Fig. 2), both in model systems and in human validation studies. The results from human omics studies can be validated at the molecular and mechanistic level by using well-defined genetic model systems that build on decades of immunological and genetic research. Applying omics approaches, for example in a specific mouse knockout condition, enables further exploration of related molecular alterations and extension of the human phenotype97. Nevertheless, mouse models do not always reflect human immunology and thus should not be used as the only means of validation. We therefore suggest the use of two or more validation strategies whenever possible. Mechanistic hypotheses can be directly evaluated within the model system with classical functional assays and extended by molecular-biology-based in vitro studies in cell culture. Insights gained from genetic model systems can then be transferred back into the human setting and further identified molecular details can be tested in the initially acquired human data sets.
Validation is possible entirely within human data sets by making use of the availability of natural variation at a locus of interest related to the identified results, that is a single nucleotide polymorphism (SNP) existing in the human population that can be studied as phenotype-linked quantitative trait loci (QTLs)53. Alternatively, genetic models can be generated in human cells through gene editing that can be assessed by functional in vitro assays, or by applying targeted CRISPR-mediated gene perturbations coupled with sequencing (Perturb-seq) that enable entire pathways or molecular networks to be targeted within a single experiment and generating omics-level readouts98.
Meta-analysis of available data sets
Another important validation approach is to link new data and findings to prior knowledge of published results (Fig. 2). Newly identified molecular phenotypes can be cross-checked in independent human studies with any classical immunological assay, such as flow cytometry or functional tests. Further, studies including omics-level information can be used to derive gene signatures for a certain cell state, a cell type or a disease, which are then tested for enrichment in the new data set. This approach is widely used in single-cell transcriptomics, as these data are ideal for generating such signatures99. Existing data are found in specialized repositories, such as the Gene Expression Omnibus (GEO), dbGAP and EGA. Sometimes, anonymized processed data, such as gene expression count data, are part of the initial publication or are available on interactive online platforms for easy access and exploration96 (https://data.humancellatlas.org/ and https://singlecell.broadinstitute.org/single_cell). One needs to know that preprocessed data might not always be ideal for secondary use, as, for example, realignment against newer versions of the genome or inclusion of sequences of pathogenic species, as well as normalization considering different covariates, are no longer possible. Under these circumstances, starting from raw data and using standardized pipelines100 is advisable. Other options to reuse existing data during validation include the investigation of newly identified genes of interest or pathways of interest in existing data sets, or the reanalysis of similar public data sets with the same algorithms as applied to the new data to identify similarities and overlap of the new findings.
Beyond comparison to existing data, the integration of newly generated data into existing data is another option for meta-analysis. This strategy is continuously performed and improved in ongoing projects of the Human Cell Atlas (HCA) consortium54. Whether data integration or validation cohorts will be the major way forward in clinical applications of omics technologies will be determined in the near future. On the basis of our experience in COVID-19 research75,101,102,103, we favor validation cohorts over data integration. While integration can be a powerful way to increase cellular resolution and enable identification of rare cellular states, it carries the risk of erroneous over-correction and loss of biological signaling. The validation-cohort approach accepts the limitation of the individual data sets but ensures the reproducibility of the observations. Both approaches have their merits and areas of application.
Validation cohorts and functional experiments
Research during the COVID-19 pandemic has taught us many principles concerning the use of omics, and particularly of single-cell multi-omics technologies. During the discovery phase, for example studying an unknown disease, single-cell multi-omics technologies provide a comprehensive overview of systemic and local changes in molecular phenotypes of affected tissues as well as the complete immune compartment. Well-defined experimental settings for clinical studies, including independent validation cohorts in combination with functional immunological validation experiments, such as flow cytometry or functional assessments of individual cell types, and potential use of animal models can lead to the discovery of cellular alterations and molecular pathways, with relevance to disease severity and trajectory and to subgroup-stratified prediction of response to potential drugs83.
Altogether, we suggest performing experiments that address molecular mechanisms and reuse of existing data as the last part of the systems immunology circle for validating the findings of a study but also for formulating subsequent hypotheses.
Conclusion
This guide is designed to provide immunologists with an entry point to use single-cell and bulk (multi-)omics technologies as a way toward a better understanding of the complex cellular and molecular interactions that operate within the immune system. This ranges from comprehensive characterization of immune homeostasis and immune variation in whole populations, the molecular definition of cell types and states, the characterization of the dynamics of immune responses, locally and systemically, to the interaction of the immune system within organ systems. Increasingly sophisticated computational algorithms, combined with perturbation experiments and ever larger data sets, enable the identification of causal relationships within data104,105,106. Furthermore, the substantially increased quality of multi-omics technologies and the high potential to standardize these technologies has already led to their application in clinical settings, paving the way towards precision medicine107. Whether it is to decipher molecular and functional mechanisms in a new disease, such as COVID-19 (refs. 75,101,102,103), to identify therapeutic targets or monitor therapeutic responses108 or whether it is to guide outcome prediction109, single-cell and bulk multi-omics technologies are suited to capture the immune system’s complexity when in action. As the omics community is well prepared for large-scale international collaborations, it is foreseeable that large scientific collaborative networks will build on the newest developments in experimental techniques as well as data-analysis approaches, which can even include concepts of specialized machine-learning approaches preserving data ownership and privacy110. We expect an enormous acceleration in knowledge once insights from multi-omics data can be used across many laboratories and institutions worldwide, without the need to share primary data.
References
Davis, M. M., Tato, C. M. & Furman, D. Systems immunology: just getting started. Nat. Immunol. 18, 725–732 (2017).
Aderem, A. Systems biology: its practice and challenges. Cell 121, 511–513 (2005).
Wagner, A., Regev, A. & Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 34, 1145–1160 (2016).
Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470 (1995).
Bennett, L. et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med 197, 711–723 (2003).
Behr, M. A. et al. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284, 1520–1523 (1999).
Xue, J. et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014).
Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
Cloonan, N. et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5, 613–619 (2008).
Carpenter, S. et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 341, 789–792 (2013).
Shalek, A. K. et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498, 236–240 (2013).
Tang, F. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Nawy, T. Single-cell epigenetics. Nat. Methods 10, 1060 (2013).
Seydel, C. Single-cell metabolomics hits its stride. Nat. Methods 18, 1452–1456 (2021).
Pai, J. A. & Satpathy, A. T. High-throughput and single-cell T cell receptor sequencing technologies. Nat. Methods 18, 881–892 (2021).
Yaari, G. & Kleinstein, S. H. Practical guidelines for B-cell receptor repertoire sequencing analysis. Genome Med. 7, 121 (2015).
Liberis, E., Velickovic, P., Sormanni, P., Vendruscolo, M. & Liò, P. Parapred: antibody paratope prediction using convolutional and recurrent neural networks. Bioinformatics 34, 2944–2950 (2018).
Miho, E. et al. Computational Strategies for dissecting the high-dimensional complexity of adaptive immune repertoires. Front. Immunol. 9, 224 (2018).
Ma, K.-Y. et al. High-throughput and high-dimensional single-cell analysis of antigen-specific CD8+ T cells. Nat. Immunol. 22, 1590–1598 (2021).
Schultheiß, C. et al. Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity 53, 442–455.e4 (2020).
Specht, H. et al. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 22, 50 (2021).
Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272 (2019).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83 (2017).
Karczewski, K. J. & Snyder, M. P. Integrative omics for health and disease. Nat. Rev. Genet. 19, 299–310 (2018).
Van den Berge, K. et al. RNA sequencing data: hitchhiker’s guide to expression analysis. Annu. Rev. Biomed. Data Sci. 2, 139–173 (2019).
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656 (2019).
Svensson, V., Vento-Tormo, R. & Teichmann, S. A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 13, 599–604 (2018).
Nakaya, H. I. et al. Systems biology of vaccination for seasonal influenza in humans. Nat. Immunol. 12, 786–795 (2011).
Rechtien, A. et al. Systems vaccinology identifies an early innate immune signature as a correlate of antibody responses to the Ebola vaccine rVSV-ZEBOV. Cell Rep. 20, 2251–2261 (2017).
Arunachalam, P. S. et al. Systems vaccinology of the BNT162b2 mRNA vaccine in humans. Nature 596, 410–416 (2021).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Gierahn, T. M. et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 (2017).
Tu, A. A. et al. TCR sequencing paired with massively parallel 3′ RNA-seq reveals clonotypic T cell signatures. Nat. Immunol. 20, 1692–1699 (2019).
Triana, S. et al. Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat. Immunol. 22, 1577–1589 (2021).
Schulte-Schrepping, J., Ferreira, H. J., Saglam, A., Hinkley, E. & Schultze, J. L. in Epigenetics of the Immune System 185–216 (Elsevier, 2020).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015).
Dimitriu, M. A., Lazar-Contes, I., Roszkowski, M. & Mansuy, I. M. Single-cell multiomics techniques: from conception to applications. Front. Cell Dev. Biol. 10, 854317 (2022).
Li, Y. et al. A functional genomics approach to understand variation in cytokine production in humans. Cell 167, 1099–1110.e14 (2016).
Gawad, C., Koh, W. & Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188 (2016).
Miller, M. B. et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022).
Miao, Z. et al. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat. Commun. 12, 2277 (2021).
Ludwig, L. S. et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339 (2019).
Mallory, X. F., Edrisi, M., Navin, N. & Nakhleh, L. Methods for copy number aberration detection from single-cell DNA-sequencing data. Genome Biol. 21, 208 (2020).
Schultze, J. L. Teaching ‘big data’ analysis to young immunologists. Nat. Immunol. 16, 902–905 (2015).
Yanai, I. & Lercher, M. A hypothesis is a liability. Genome Biol. 21, 231 (2020).
Brusic, V., Gottardo, R., Kleinstein, S. H. & Davis, M. M. & HIPC steering committee. Computational resources for high-dimensional immune analysis from the Human Immunology Project Consortium. Nat. Biotechnol. 32, 146–148 (2014).
De Jager, P. L. et al. ImmVar project: Insights and design considerations for future studies of ‘healthy’ immune variation. Semin. Immunol. 27, 51–57 (2015).
Ter Horst, R. et al. Host and environmental factors influencing individual human cytokine responses. Cell 167, 1111–1124 (2016).
Thomas, S. et al. The Milieu Intérieur study—an integrative approach for study of human immunological variance. Clin. Immunol. 157, 277–293 (2015).
Schultze, J. L., SYSCID consortium & Rosenstiel, P. Systems medicine in chronic inflammatory diseases. Immunity 48, 608–613 (2018).
Momozawa, Y. et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 9, 2427 (2018).
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
Plichta, D. R., Graham, D. B., Subramanian, S. & Xavier, R. J. Therapeutic opportunities in inflammatory bowel disease: mechanistic dissection of host-microbiome relationships. Cell 178, 1041–1056 (2019).
Reyes, M. et al. An immune-cell signature of bacterial sepsis. Nat. Med. 26, 333–340 (2020).
Su, Y. et al. Multi-omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell 183, 1479–1495 (2020).
Schmiedel, B. J. et al. Single-cell eQTL analysis of activated T cell subsets reveals activation and cell type-dependent effects of disease-risk variants. Sci. Immunol. 7, eabm2508 (2022).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Frishberg, A. et al. Multiple trajectory alignment reconstructs disease dynamics for discovery and clinical benefit. Cell Rep. Med. https://doi.org/10.1016/j.xcrm.2022.100652 (2022).
Querec, T. D. et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10, 116–125 (2009).
Wimmers, F. et al. The single-cell epigenomic and transcriptional landscape of immunity to influenza vaccination. Cell 184, 3915–3935.e21 (2021).
McShane, L. M. et al. Criteria for the use of omics-based predictors in clinical trials. Nature 502, 317–320 (2013).
Tung, P.-Y. et al. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 7, 39921 (2017).
Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020).
McGinnis, C. S. et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019).
Kang, H. M. et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018).
De Jong, S. et al. Seasonal changes in gene expression represent cell-type composition in whole blood. Hum. Mol. Genet. 23, 2721–2728 (2014).
Adrover, J. M. et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity 50, 390–402 (2019).
Temba, G. S. et al. Urban living in healthy Tanzanians is associated with an inflammatory status driven by dietary and metabolic changes. Nat. Immunol. 22, 287–300 (2021).
Denisenko, E. et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21, 130 (2020).
Haque, A., Engel, J., Teichmann, S. A. & Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 75 (2017).
Massoni-Badosa, R. et al. Sampling time-dependent artifacts in single-cell genomics studies. Genome Biol. 21, 112 (2020).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Schulte-Schrepping, J. et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440.e23 (2020).
van der Wijst, M. G. P. et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 13, eabh2624 (2021).
Kim, H. H., Park, Y. R., Lee, K. H., Song, Y. S. & Kim, J. H. Clinical MetaData ontology: a simple classification scheme for data elements of clinical data based on semantics. BMC Med Inf. Decis. Mak. 19, 166 (2019).
Baruzzo, G. et al. Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 14, 135–139 (2017).
Schmid, K. T. et al. scPower accelerates and optimizes the design of multi-sample single cell transcriptomic studies. Nat. Commun. 12, 6625 (2021).
Vieth, B., Ziegenhain, C., Parekh, S., Enard, W. & Hellmann, I. powsimR: power analysis for bulk and single cell RNA-seq experiments. Bioinformatics 33, 3486–3488 (2017).
Zappia, L. & Theis, F. J. Over 1000 tools reveal trends in the single-cell RNA-seq analysis landscape. Genome Biol. 22, 301 (2021).
Anders, S. et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786 (2013).
Aschenbrenner, A. C. et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med 13, 7 (2021).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Lähnemann, D. et al. Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020).
Reimand, J. et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14, 482–517 (2019).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Chu, X. et al. Integration of metabolomics, genomics, and immune phenotypes reveals the causal roles of metabolites in disease. Genome Biol. 22, 198 (2021).
Argelaguet, R. et al. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 14, e8124 (2018).
Rautenstrauch, P., Vlot, A. H. C., Saran, S. & Ohler, U. Intricacies of single-cell multi-omics data integration. Trends Genet. 38, 128–139 (2022).
Li, R., Li, L., Xu, Y. & Yang, J. Machine learning meets omics: applications and perspectives. Brief. Bioinforma. 23, bbab460 (2022).
Lotfollahi, M. et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 40, 121–130 (2022).
Wilkinson, M. D. et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Scholz, C. J. et al. FASTGenomics: an analytical ecosystem for single-cell RNA sequencing data. Preprint at bioRxiv https://doi.org/10.1101/272476 (2018).
Bonaguro, L. et al. CRELD1 modulates homeostasis of the immune system in mice and humans. Nat. Immunol. 21, 1517–1527 (2020).
Przybyla, L. & Gilbert, L. A. A new era in functional genomics screens. Nat. Rev. Genet. 23, 89–103 (2022).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Collado-Torres, L. et al. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 35, 319–321 (2017).
Krämer, B. et al. Early IFN-α signatures and persistent dysfunction are distinguishing features of NK cells in severe COVID-19. Immunity 54, 2650–2669.e14 (2021).
Bernardes, J. P. et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity 53, 1296–1314.e9 (2020).
Georg, P. et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 185, 493–512.e25 (2022).
Dixit, A. et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell rna profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Datlinger, P. et al. Ultra-high-throughput single-cell RNA sequencing and perturbation screening with combinatorial fluidic indexing. Nat. Methods 18, 635–642 (2021).
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896.e15 (2016).
De Domenico, E. et al. Optimized workflow for single-cell transcriptomics on infectious diseases including COVID-19. STAR Protoc. 1, 100233 (2020).
Cohen, Y. C. et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 27, 491–503 (2021).
van Galen, P. et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265–1281.e24 (2019).
Warnat-Herresthal, S. et al. Swarm Learning as a privacy-preserving machine learning approach for disease classification. Preprint at bioRxiv https://doi.org/10.1101/2020.06.25.171009 (2020).
Mair, F. et al. A targeted multi-omic analysis approach measures protein expression and low-abundance transcripts on the single-cell level. Cell Rep. 31, 107499 (2020).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
De Simone, M., Rossetti, G. & Pagani, M. Single cell T cell receptor sequencing: techniques and future challenges. Front. Immunol. 9, 1638 (2018).
Zhang, W. et al. A framework for highly multiplexed dextramer mapping and prediction of T cell receptor sequences to antigen specificity. Sci. Adv. 7, eabf5835 (2021).
Guo, H. et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 23, 2126–2135 (2013).
Farlik, M. et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 10, 1386–1397 (2015).
Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007).
Rotem, A. et al. Single-cell ChIP–seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172 (2015).
Wu, F., Olson, B. G. & Yao, J. DamID-seq: Genome-wide mapping of protein-dna interactions by high throughput sequencing of adenine-methylated DNA Fragments. J. Vis. Exp. e53620 (2016).
Kind, J. et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147 (2015).
Bartosovic, M., Kabbe, M. & Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021).
Schones, D. E. et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 (2008).
Gao, W., Lai, B., Ni, B. & Zhao, K. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq. Nat. Protoc. 15, 68–85 (2020).
Kelly, T. K. et al. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506 (2012).
Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 6, e23203 (2017).
Song, L. & Crawford, G. E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, pdb.prot5384 (2010).
Buenrostro, J. D. et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548 (2018).
Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019).
Cusanovich, D. A. et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324 (2018).
Belton, J.-M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Ramani, V. et al. Sci-Hi-C: a single-cell Hi-C method for mapping 3D genome organization in large number of single cells. Methods 170, 61–68 (2020).
Shendure, J. et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017).
McKinnon, K. M. Flow cytometry: an overview. Curr. Protoc. Immunol. 120, 5.1.1–5.1.11 (2018).
Cheung, P. et al. Single-cell chromatin modification profiling reveals increased epigenetic variations with aging. Cell 173, 1385–1397 (2018).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Katzenelenbogen, Y. et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in Cancer. Cell 182, 872–885 (2020).
Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 12, 668–679 (2016).
Li, Z. et al. Single-cell lipidomics with high structural specificity by mass spectrometry. Nat. Commun. 12, 2869 (2021).
Davis, S. et al. Seandavi/Awesome-Single-Cell: 2018-06-20-1. Zenodo https://doi.org/10.5281/zenodo.1294021 (2018).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Stevens, I. et al. Ten simple rules for annotating sequencing experiments. PLoS Comput. Biol. 16, e1008260 (2020).
Vita, R., Overton, J. A., Mungall, C. J., Sette, A. & Peters, B. FAIR principles and the IEDB: short-term improvements and a long-term vision of OBO-foundry mediated machine-actionable interoperability. Database 2018, bax105 (2018).
Acknowledgements
J.L.S. is supported by the German Research Foundation (DFG) under Germany’s Excellence Strategy (EXC2151-390873048), as well as under SCHU 950/8-1; GRK 2168, TP11; SFB704, the BMBF-funded excellence project Diet–Body–Brain (DietBB); and the EU project SYSCID under grant number 733100. A.C.A. is supported by DFG under AS 637/1-1; AS 637/2-1; AS 637/3-1 and SFB1454/P02 (project no. 432325352). M.B. is supported by DFG (IRTG2168-272482170, SFB1454-432325352).
Author information
Authors and Affiliations
Contributions
L.B., J.S.-S. and J.L.S. developed the concept. L.B., J.S.-S., T.U., M.B., A.C.A. and J.L.S. discussed the concept. L.B. and J.L.S. designed the figures. L.B., J.S.-S., T.U., M.B. and J.L.S. wrote the original draft. L.B., J.S.-S., T.U., M.B., A.C.A. and J.L.S. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks Uri Hershberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. N. Bernard was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bonaguro, L., Schulte-Schrepping, J., Ulas, T. et al. A guide to systems-level immunomics. Nat Immunol 23, 1412–1423 (2022). https://doi.org/10.1038/s41590-022-01309-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-022-01309-9
This article is cited by
-
Integrating single-cell multi-omics and prior biological knowledge for a functional characterization of the immune system
Nature Immunology (2024)
-
Locally sourced: site-specific immune barriers to metastasis
Nature Reviews Immunology (2023)
-
Transforming bioengineering with unbiased teams and tools
Nature Reviews Bioengineering (2023)
