
Abstract
Ten years since the immune checkpoint inhibitor ipilimumab was approved for advanced melanoma, it is time to reflect on the lessons learned regarding modulation of the immune system to treat cancer and on novel approaches to further extend the efficacy of current and emerging immunotherapies. Here, we review the studies that led to our current understanding of the melanoma immune microenvironment in humans and the mechanistic work supporting these observations. We discuss how this information is guiding more precise analyses of the mechanisms of action of immune checkpoint blockade and novel immunotherapeutic approaches. Lastly, we review emerging evidence supporting the negative impact of melanoma metabolic adaptation on anti-tumor immunity and discuss how to counteract such mechanisms for more successful use of immunotherapy.
Similar content being viewed by others

Main
Melanoma is the most aggressive and deadly of skin cancers. Cutaneous melanomas are the most frequent, and are typically associated with ultraviolet (UV) exposure and elevated tumor mutational burden (TMB), which contribute to high immunogenicity. Several historical observations point to melanoma as an immune-responsive tumor. First, 5% of patients with melanomas present with metastatic disease that genotypically resembles cutaneous melanoma, but without an identifiable primary melanoma, suggesting that the primary tumor may have spontaneously regressed1. Second, melanoma is often associated with vitiligo—the manifestation of an autoimmune reaction against melanocytes, indicative of cross-reactive immune responses targeting melanoma and normal melanocytes. Vitiligo was shown to be a favorable prognostic indicator in patients with melanoma2, suggesting that anti-melanocytic immune responses help control melanoma growth. Third, melanomas can be infiltrated by reactive lymphocytes3, with dense infiltration of peritumoral lymphocytes being associated with better prognosis. Melanoma classification based on tumor-infiltrating lymphocyte (TIL) distribution (brisk, non-brisk, and absent) is still used today4,5,6,7. However, melanoma disseminates and metastasizes very easily, indicating that active immune suppression or dysfunction must offset its immunogenicity. Reliance on immune-evasion mechanisms for disease progression may underscore the specific vulnerability of melanoma to immunotherapy, thus explaining its unique responsiveness to these treatments.
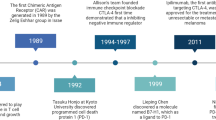
The concept of immune checkpoint blockade (ICB) for the treatment of cancer was pioneered by Jim Allison and colleagues, who showed that antibodies blocking the T cell co-inhibitory receptor CTLA-4 can regress tumors in mice8. Human CTLA-4 blocking antibodies were then developed and tested in cancer patients, with ipilimumab becoming the first therapy to extend survival in metastatic melanoma9,10, which led to its approval for treating this disease in 2011. PD-1 was recognized as another key T cell immune checkpoint11. PD-1 or PD-L1 blocking antibodies have been found to enhance tumor control in mice12,13 and CD8+ T cell functionality in a chronic viral infection model14. Promising results in early clinical trials with PD-1 blocking antibodies in refractory solid tumors were confirmed in phase 3 studies in melanoma, where the PD-1 inhibitors pembrolizumab and nivolumab were found to extend survival compared with ipilimumab or chemotherapy15,16,17,18,19. These agents were then approved for the treatment of metastatic melanoma in 2014.
Overall, the clinical success with ICB in melanoma has confirmed the therapeutic impact of re-invigorating the immune system to effectively target this disease. However, even in the optimal scenarios with combination ICB, approximately half of patients do not derive long-lasting benefit20. This indicates the need for better predictive biomarkers of response and new rational targets for more effective combination treatments to overcome immune resistance. While elevated tumor PD-L1 expression and TMB have been found to correlate with clinical responses to ICB in melanoma21, these biomarkers cannot accurately predict outcomes in all cases. Because the longest and most consolidated clinical experience with ICB is in melanoma, this information can be now leveraged to achieve a more precise understanding of the molecular determinants of activity of these therapies in people.
Here, we provide an updated overview of the immune landscape of human melanoma and how it is modulated by ICB, focusing on studies in humans. Moreover, we highlight current limitations of immunotherapy and delineate the next potential avenues to improve the use of these treatments.
Melanoma-specific T cells and their therapeutic potential
Melanoma TILs are enriched for specificity to melanoma-associated antigens, indicating that anti-melanoma T cells can undergo priming, expansion, and recruitment to the tumor (Fig. 1a). Endogeneous T cell responses to melanoma have been exploited for multiple objectives, including (1) identification of the cognate antigens that can then be used for vaccine development and (2) expansion and/or engineerization of tumor-specific T cells for adoptive cell therapy (ACT).

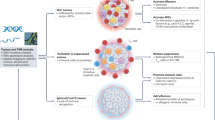
a, Schematic representation of the principal immune components in the melanoma microenvironment. Four main functional modules can be distinguished: (1) a CD8 module (red/orange/yellow), including cytotoxic T cells (CTLs) and a spectrum of dysfunctional cells including progenitor TEX (pTEX) and terminal TEX cells; (2) an innate module (violet/purple) that can impact the recruitment and activation of T cells depending on the tolerogenic (tolerogenic DCs; myeloid-derived suppressor cells, MDSC), pro-tumor (M2), or anti-tumor (DCs) inflammatory potential; (3) a CD4 module (green), which is highly heterogeneous and can be shaped by the immune cells in the other modules and includes immunosuppressive Treg cells, TH2 cells with pro-tumor inflammatory potential, and TFH-like cells, which can promote B cell function; and (4) a B cell module (blue), including B cells in various stages of differentiation up to plasma cells (PCs), with pro- or anti-tumor function depending on the profile of immunoglobulins produced (IgA, IgG2, IgG4 versus IgG1, respectively) and expression of co-inhibitory molecules (for example IL-10 and PD-L1). Each of these immune modules has a counter-regulatory program to dampen immune responses, thus explaining the coexistence of tumor cells and anti-tumor lymphocytes in the same environment. b, The immune infiltrate in melanoma can organize in cellular aggregates defined as tertiary lymphoid structures (TLSs), with B cells at the core surrounded by T cells and APCs, which generate a germinal-center-like pattern. The impact of these structures on anti-tumor immunity is likely determined by the potential to recruit or expand CTLs and TH1 cells versus immunosuppressive or TEX cells.
The easy access to cutaneous melanoma lesions prompted the study of melanoma TILs, which was further facilitated by the discovery of IL-2 (ref. 22) in 1977, enabling expansion of T cells in vitro for their characterization. T cells recognizing melanocyte differentiation antigens (for example MelanA and gp100), cancer germline (cancer-testis) antigens, and tumor-overexpressed antigens in human melanoma were then identified (Box 1) and comprehensively profiled using peptide–MHC multimers23,24. More recently, high-throughput antigen screening coupled with next-generation sequencing of both T cell receptor (TCR) repertories and tumor genomes have substantially expanded our understanding of anti-tumor T cell specificities and their dynamics25. Indeed, advances in tumor sequencing and associated computational approaches have allowed the identification of tumor-mutation-derived neoantigens (Box 1) that contribute to tumor immunogenicity and T cell recognition25. Neoantigens are particularly abundant in melanoma, given the elevated frequency of somatic mutations in this disease26. Notably, many anti-melanoma TILs recognize neoantigens derived from tumor somatic mutations27,28,29, which are attractive for tumor-specific targeting and reduced autoimmune toxicity. To boost T cell responses in vivo against tumor neoantigens, vaccines with tumor-associated neoepitopes have been demonstrated to be safe and to elicit potent tumor-specific T cell responses in patients with melanoma30,31,32. These responses included both CD4+ and CD8+ T cells, pointing towards dual CD4+ and CD8+ immunogenicity of strong neoepitopes and possibly to the ability of CD4+ T cells to directly control melanoma tumors. This mechanism may be particularly relevant in the setting of melanoma, considering its unique MHC-II expression.
Overall, the existence of melanoma-specific TILs indicated that in many cases (1) these cells are numerically or functionally insufficient to completely eradicate the tumor, and (2) they could be boosted to achieve the numbers required for complete tumor eradication in vivo. Toward this goal, ACT using ex vivo-expanded TILs has been shown to contribute to tumor regressions, especially in melanoma. The Rosenberg group at the Surgery Branch of the National Cancer Institute and other teams demonstrated that ACT of TILs can be clinically effective, further confirming the tumor reactivity of melanoma-derived T cells33,34,35,36. Interestingly, ACT using TIL products enriched for tumor-neoantigen specificities were found to produce durable clinical responses with no significant toxicity37,38,39,40,41,42, suggesting a potential role for T cells recognizing tumor-associated neoantigens in the activity of ACT.
From the early to the most advanced characterization of melanoma TIL specificities and the use of these cells as anti-cancer therapy, these studies have extensively corroborated the biologic and therapeutic relevance of T cell responses in patients with melanoma. Whether the recent developments with neoantigen-specific immunotherapy will lead to the next chapter of ACT and/or vaccines in melanoma remains to be determined, especially considering that neoantigens arise from unique tumor-specific mutations rarely shared between indidividuals. Thus, such interventions would require patient-specific identification and product manufacturing25.
B cell responses in melanoma
Melanoma antigens can also elicit B cell responses, further supporting the immunogenicity of this disease. Serological analyses from patients wih melanoma contributed to the definition of several melanoma-associated antigens43,44,45. Autoantibodies against melanoma-associated antigens have been reported to develop in patients with melanoma and to correlate with improved prognosis in certain cases46,47,48. However, whether anti-melanoma antibody responses play a causal role in tumor protection remains to be fully elucidated. Mature B cells can be found in melanoma lesions at higher frequencies compared with normal skin, and have been described to localize in aggregates with T cells and dendritic cells (DCs), defined as tertiary lymphoid structures (TLSs)49,50 (Fig. 1b). TLSs are ectopic lymphoid structures that typically form in response to chronic inflammation and evolve dynamically to adapt to the local tissue injury51. Mature TLSs comprising T and B cells in germinal-center-like zones are found more frequently than immature, B-cell-depleted TLSs in metastatic melanoma lesions49. Key mediators of TLS formation, including early (CXCL13), intermediate (lymphotoxin beta receptor), and later lympho-angiogenic factors (CCL21, LIGHT), can be overexpressed in metastatic melanoma52,53. It is possible that chronic immunogenic stimuli elicited by melanoma-associated antigens trigger TLS-supporting signals that recruit and expand tumor-specific B cells. In TLS, B cells can theoretically undergo maturation into antibody-secreting cells in the presence of proper T cell-mediated helper signals and/or can serve as antigen-presenting cells (APCs) that contribute to local tumor-specific T cell priming. The relative impact of these two B cell functions in TLSs, as well as the role of B cell antigen specificity and functionality in tumors, remains to be precisely and comprehensively elucidated. B cells and their antibody products may be highly heterogeneous, with profiles ranging from pro-inflammatory (IgG1+) to immunosuppressive (for example CD1d+IL-10+PD-L1+, IgA+, IgG2+, or IgG4+) (Fig. 1b). This heterogeneity may explain the apparently conflicting results from independent studies reporting associations between B cell infiltrates and either favorable50,54,55 or negative56 prognosis in people with melanoma. These discrepancies may be also explained by the variety of microenvironmental immune signals and their effects on intratumor B cell polarization toward pro- or anti-inflammatory profiles. TLSs are highly dynamic and can also attract immunosuppressive cells, such as regulatory T (Treg) cells, immature tolerogenic DCs, and/or myeloid-derived suppressor cells (MDSCs), in response to excessive inflammation57,58,59 (Fig. 1b). It is still not clear whether bona fide T follicular helper (TFH) cells with germinal center B cell-selective features can differentiate in tumor-associated TLSs. However, T cells with a similar phenotype have been found to overlap with dysfunctional and/or non-conventional suppressive T cells in chronic viral infection and tumors60,61, suggesting their potential negative impact on anti-tumor immunity. The possibility of precisely measuring the immune stimulatory versus immunosuppressive potential of tumor-associated TLSs, and to predict their fate based on composition and inflammatory signals, would offer a potentially valuable biomarker for response to immunotherapy. Indeed, despite the many unknowns in TLS and intratumor B cell biology, these factors have been recently reported to associate with improved tumor responses to ICB in patients62,63,64. These initial findings open new avenues to potentially improve immunotherapy by dissecting the biology, heterogeneity, and mechanistic role of TLSs in tumors and find ways to properly manipulate these structures for potentiating local anti-tumor immune responses. Mechanistic studies in this space are limited by the lack of TLS formation in mouse tumor models, restricting these analyses to correlative observations in patients. Engineering animal models to induce spontaneous TLSs in tumors could help add causal inference to these correlative observations and facilitate an understanding of how the manipulation of these structures alters B cell versus T cell responses and the outcome to different immunotherapies.
Immunosuppressive mechanisms in melanoma
Despite the immunogenicity of melanoma, metastatic melanoma is generally not eliminated spontaneously. The strong immune selective pressure in response to melanoma immunogenicity may induce the tumor to adapt and suppress anti-tumor immunity. In addition, local inflammation can activate homeostatic immunologic feedback, which contributes to this adaptive resistance. As an example, intratumoral CD8+ T cells, by producing CCL22 and IFN-γ, induce intratumor Treg accumulation and PD-L1 on tumor cells, respectively65,66. In turn, melanoma-specific TILs are functionally hampered in human melanoma lesions but can regain functionality after ex vivo culture with the proper cytokine growth factors67.
By binding PD-1 expressed on tumor-specific T cells, PD-L1 and PD-L2 induce a negative signaling cascade downstream of PD-1, which dampens T cell activation and tumoricidal function11. Therefore, in melanoma cells, expression of PD-L1 or PD-L2 can offset the positive T cell signals delivered by both MHC-I and MHC-II antigen-presentation routes (Box 2).
Melanoma can also directly attract immune inhibitory cells. Especially through MHC-II expression, melanoma cells have a unique capacity to interact with and attract immunosuppressive CD4+ T cell subsets. Treg cells are increased in peripheral blood (PB), lymph nodes, and in the tumor microenvironment (TME) of patients with melanoma, and have been found to inhibit TIL function61,68,69. The typical Treg increase in metastatic lymph nodes suggests that melanoma may directly modulate its microenvironment to evade immune surveillance and grow (Fig. 1a). In early studies, melanoma-associated Treg cells were found to recognize melanoma antigens70,71. This has been recently confirmed by initial single-cell omics analyses showing that melanoma-infiltrating Treg cells are highly clonal and can recognize tumor cells via TCR–pMHC-II interactions, suggesting that melanoma cells can directly activate and expand Treg cells, thus controlling local immunosuppression72. Notably, clonal expansion of tumor-specific Treg cells has been found to be associated with neoantigen burden, which in turn correlated with tumor expression of MHC-II, further pointing to a mechanism whereby melanoma controls Treg expansion via MHC-II expression depending on its immunogenicity72.
Moreover, in patients with melanoma, a skew toward type 2 T helper (TH2)-polarized CD4+ T cells and related cytokines has been reported, once again highlighting an altered CD4+ T cell compartment in the presence of melanoma73,74,75. A TH2 immune bias is an indicator of chronic and counterproductive inflammatory responses. Over-production of VEGF and galectin-9 (GAL9) by human melanoma cells can support such TH2 bias via M2 macrophage differentiation, thus contributing to tumor-promoting inflammation73,75 (Fig. 1a).
Suboptimal co-stimulation due to insufficient mature DCs may also contribute to a hostile melanoma microenvironment for T cells. DCs play a key role in controlling local inflammation, T cell recruitment, and activation in melanoma76,77,78,79 (Fig. 1a). In mouse melanoma models with a constitutively active beta-catenin signaling pathway, lack of infiltrating antigen cross-presenting CD103+ DCs contributed to T cell exclusion from the TME80,81. These observations underscore two important points: (1) active priming in the TME is key to maintaining the local T cell pool and to preserving TIL tumoricidal function and (2) the tumor pro-oncogenic program can directly affect DC recruitment, priming capacity, and costimulatory potential in the TME. Similarly, abnormal production of prostaglandin E2 (PGE2) by melanoma cells has been shown to limit intratumoral recruitment of cross-presenting DC subsets, possibly via downregulation of the DC chemoattractants XCL1 and CCL5 in intratumor natural killer cells82. Importantly, in cutaneous melanoma, UV exposure has been shown to promote the tolerogenic profile of a specialized subset of epidermal DCs called Langerhans cells, which sense the skin barrier surface through langerin—a C-type lectin that functions as a pattern recognition receptor83,84. These cells can efficiently migrate to the skin-draining lymph nodes. Upon UV exposure, suboptimally activated immature Langerhans cells migrate into the draining lymph nodes, tolerizing T cells in an antigen-specific way (Box 2). However, some controversy has emerged with respect to the specific type of langerin-expressing DCs primarily contributing to this tolerogenic phenotype84,85. A homeostatic/tolerogenic program was also found to be induced by IFN-γ in DCs infiltrating human tumors, including melanoma86. Overall, these observations indicate that melanoma cells and/or local inflammation, via multiple mechanisms, can negatively affect intratumor DC abundance and costimulatory capacity, thus limiting the generation of potent T cell responses. Considering the plasticity of DCs, the same findings would point to new potential therapeutic opportunities by re-polarizing DC function and restoration of sufficient local anti-tumor T cell priming and activation87.
Recent progress with ICB therapy in melanoma
As detailed above, the immunogenicity of melanoma—demonstrated by the presence of specific adaptive immune responses—can be offset by activation of regulatory programs that would normally serve to prevent immune pathology. As part of this regulation, immune checkpoint molecules are expressed on activated antigen-experienced TILs and have been found to represent effective immunotherapeutic targets in melanoma. CTLA-4 was the first immune checkpoint to be identified. CTLA-4 on T cells competes with the costimulatory molecule CD28 for the same ligands, CD80 and CD86. However, CTLA-4 binds CD80 and CD86 with greater affinity and avidity than does CD28 (ref. 88), depriving T cells of costimulatory signals. These discoveries led to the seminal finding that CTLA-4 blocking antibodies can regress tumors in mice8, followed by the clinical development of these reagents, with ipilimumab obtaining first indication in metastatic melanoma in 2011 (refs. 9,10) (Table 1). PD-1 was the second immune checkpoint to be discovered11. CD8+ exhausted T (TEX) cells progressively lose effector functions upon chronic antigen stimulation during infection and cancer and overexpress PD-1. PD-1 or PD-L1 blockade can reinvigorate these cells, resulting in their numeric expansion and restoration of effector functions in mice14,89 and humans90,91,92. These effects were associated with enhanced tumor control in preclinical models12,13,93, which translated into substantial therapeutic activity in cancer patients, with the PD-1 inhibitors pembrolizumab and nivolumab being approved in metastatic melanoma in 2014 (refs. 15,16,17,18) (Table 1). Considering the different and potentially complementary effects of CTLA-4 and PD-1 blockade94, these therapies were subsequently tested in combination, demonstrating greater long-term efficacy than either agent alone in metastatic melanoma, with 49% of treated individuals remaining alive after 6.5 years (Table 1), albeit at the cost of greater toxicity20. Alternative regimens and/or dosages of anti-PD-1 + anti-CTLA-4 are currently being investigated to reduce toxicity95.
The discovery of immune co-inhibitory receptors and their co-expression in TEX cells89 has inspired further development of antibody therapeutics targeting novel immune checkpoint molecules. The most promising emerging ICB target is LAG-3. LAG-3 is a surface inhibitory receptor that has structural similarity to CD4 and competitively binds to MHC-II and other ligands, including galectin-3. Among other cell types, LAG-3 is upregulated on antigen-stimulated T cells, including TEX cells. Similar to CTLA-4, LAG-3 is also constitutively overexpressed on Treg cells, contributing to their suppressive function96. While LAG-3 blockade as monotherapy has modest anti-tumor efficacy, combined anti-LAG-3 + anti-PD-1 exhibited substantially enhanced therapeutic activity in several mouse tumor models, including models of melanoma96. Several antibody therapeutics targeting LAG-3 by mainly blocking the MHC-II–LAG-3 interactions are being tested in individuals with cancer96. The most advanced anti-LAG-3 antibody is relatlimab, which has been tested in combination with nivolumab in a phase 2–3 clinical trial in participants with previously untreated metastatic or unresectable melanoma. Results from this trial indicate that progression-free survival (PFS) following relatlimab + nivolumab treatment is greater than PFS following nivolumab monotherapy and similar to PFS following ipilimumab + nivolumab historically97 (Table 1). Increases in circulating LAG-3+ T cells correlate with shorter survival and/or disease progression after PD-1 blockade in patients with melanoma98,99, supporting the rationale for combined PD-1 and LAG-3 inhibition. Whether anti-PD-1 + anti-LAG-3 has comparable efficacy to anti-CTLA-4 + anti-PD-1 and decreased toxicity, and whether circulating LAG-3+ T cells can serve as a biomarker to allocate patients to anti-LAG-3-containing therapies, remain to be directly tested in prospective clinical trials. Similarly, it will be important to understand whether MHC-II expression levels and localization play a role in the activity of LAG-3 blockade.
Additional advancements with ICB in melanoma have come from its investigation in earlier-stage disease, when administered after (adjuvant therapy) or before (neoadjuvant therapy) surgical resection. Ipilimumab was the first ICB therapy to show durable survival benefit in melanoma in the adjuvant setting100,101,102 (Table 1), followed by PD-1 blockade with either nivolumab or pembrolizumab, which showed improved relapse-free survival (RFS) compared with placebo or even ipilimumab in individuals with high-risk stage-III melanoma, although no overall survival (OS) benefit has been observed to date103. Given the improved toxicity profile compared with that of ipilimumab, PD-1 blockade has become a standard of care for adjuvant therapy. Recently, adjuvant pembrolizumab received Food & Drug Administration approval for stage-IIb/c melanoma on the basis of its improved RFS compared with that of placebo104.
Neoadjuvant ICB has also gained momentum, with five studies being completed to date in melanoma105,106. Neoadjuvant ipilimumab + nivolumab or PD-1 blockade alone demonstrated 33–57% and 19–25% complete pathologic response (pCR) rates, respectively. As the increased pCR rate with neoadjuvant ipilimumab + nivolumab comes at the cost of greater toxicity, studies to optimize doses and/or regimens for a better safety profile are underway (Table 1). Alternative combination regimens are also being investigated, with neoadjuvant nivolumab + relatlimab showing impressive pCR rates (59%)107.
Notably, early pathologic responses to neoadjuvant ICB strongly predict RFS. Pathologic responses (<50% viable tumor) within 6 weeks of anti-PD-1 ± anti-CTLA-4 were associated with >94% RFS at 2 years in a recent pooled analysis of individuals with stage III melanoma108. The ability to predict long-term outcomes on the basis of early pathological responses to neoadjuvant therapy provides the unique ability to tailor the type and duration of adjuvant therapy in a personalized manner. Moreover, initial biomarker analyses have reported that elevated baseline IFN-γ signature scores and TMB as well as post-treatment tumor fibrosis are associated with pathologic responses and survival after neoadjuvant ICB109,110, offering biological parameters that can further improve patient allocation to the right treatment. Trials that test whether these potential biomarkers can be used to inform clinical management are underway. For example, ongoing trials are investigating the advantage of modifying adjuvant therapy (NCT04013854) or surgical management111 on the basis of pathologic response to neoadjuvant therapy or baseline tumor IFN-γ scores (NCT4133948). Finally, the clinical efficacy of neoadjuvant versus adjuvant ICB with pembrolizumab is being directly tested in prospective studies (NCT03698019, Table 1).
Overall, these studies point to the feasibility of ICB in earlier-stage disease. In particular, neoadjuvant ICB offers the advantage of rapid activity evaluation, which facilitates early treatment modification if necessary. Moreover, neoadjuvant treatment serves as an efficient platform for analysis of immune correlates and resistance mechanisms in patients, which is key to guide the most rational treatment combinations, as discussed below.
Assessment of pharmacodynamic ICB activity for refined biomarker discovery
The precise tracking and interrogation of the immunologic responses to ICB are critical for understanding the mechanism(s) of action and identifying early predictive biomarkers. The direct pharmacodynamic effect of immunotherapy is on the immune compartment, with an indirect effect on the tumor. Neoadjuvant studies offer an optimal setting to investigate response predictors and tumor resistance mechanisms in humans because of the availability of paired pre-treatment and on-treatment samples. Early neoadjuvant trials in melanoma identified pharmacodynamic immune responses of CD4+ and CD8+ TILs to high-dose IFN alpha-2b112 and anti-CTLA-4 (ref. 106) at 4–6 weeks post therapy, demonstrating that the immunologic activity of immunotherapies occurs early in humans.
Subsequent studies interrogated changes in cellular states in response to ICB in more depth. Through these analyses, CTLA-4 blockade was found to primarily expand ICOS+T-bet+CD4+ effector T cells in PB and tumors, including melanoma113,114,115. Treg cells have been extensively studied as an another potentially relevant pharmacodynamic target of CTLA-4 blockade, because CTLA-4 is maximally expressed in Treg cells, especially intratumoral Treg cells116. While in murine tumor models, including melanoma, intratumoral Treg cells selectively decrease after anti-CTLA-4 and this contributes to the anti-tumor activity (Fig. 2), similar effects have not been definitely proven in humans. Ipilimumab is an IgG1 antibody and can mediate antibody-dependent-cellular-cytotoxicity (ADCC) of human Treg cells ex vivo117. However, Foxp3+ Treg cells were not found to be reduced in melanoma biopsies after ipilimumab treatment118. In addition, in mice and humans, peripheral Treg cells are overall expanded after CTLA-4 blockade independent of the therapeutic activity106,119,120. This indicates that CTLA-4 can limit Treg expansion and that when this pathway is blocked more Treg cells are generated in the periphery, despite potentially being less suppressive during CTLA-4 inhibition (Fig. 2)61,121,122. This peripheral Treg expansion can contribute to replenish the intratumor Treg pool, thus explaining our inability to detect substantial intratumoral Treg loss even upon treatment with a depleting anti-CTLA-4 antibody. Deeper Treg profiling will help us understand whether certain Treg subsets (for example CTLA-4hi Treg cells) may be specifically modulated and/or reduced by CTLA-4 blockade and can be monitored as robust biomarkers. There may also be distinct mechanisms of action of anti-CTLA-4 against peripheral and intratumoral Treg cells, which can be determined by the different composition and function of the microenvironment. These observations led to the development of optimized versions of the anti-CTLA-4 ipilimumab for increased ADCC of Treg cells or preferential activation in the TME to limit the effect on peripheral Treg cells123. These agents are currently in clinical testing for advanced solid cancers, including melanoma.

Schematic representation of the immune cell types primarily affected by anti-CTLA-4 (red arrows) and anti-PD-1 (blue arrows) as reported in humans. Notably, most of these cell types overexpress the targets of ICB, potentially explaining their preferential modulation after ICB. CTLA-4 blockade induces TH1 effector cells (eff) and TFH cells, while counteracting Treg function and possibly expanding TEX cells. PD-1 blockade reinvigorates TEX and possibly potentiates effector CD8+ CTLs but expands functionally suppressive Treg cells and can decrease TFH cells. Combined assessment of these immune cell subsets during ICB will likely help select relevant pharmacodynamic changes that can be then taken as reliable activity biomarkers for these treatments. Dark color, definitive evidence; light color, weaker evidence.
Recent studies have also carefully profiled the kinetics and composition of the pharmacodynamic immune responses of human cancers—including melanoma—to PD-1 blockade. PD-1 blockade results in early activation and proliferation of T cells and Treg cells. The CD8 response is largely composed of TEX over-expressing PD-1, CTLA-4, and Eomes90,91. Early CD8+ TEX reinvigoration was associated with clinical and pathologic responses after a single dose of PD-1 blockade, with TEX proliferation peaking by 7 days after the initial treatment dose90. The heterogeneity of CD8+ TEX cells is now being deconvoluted, with specific TEX subsets shown to preferentially respond to anti-PD-1, such as progenitor CD8+ TEX cells124. However, the immunologic response to PD-1 blockade in humans is heterogenous and may extend to other CD8+ T cell subsets, such as effector-memory CD8+ T cells125 (Fig. 2). Increases in Treg proliferation and activation after PD-1 blockade were associated with poor clinical outcomes90,91. These data are consistent with recent studies showing that PD-1 blockade potentiates Treg immunosuppression126. Therefore, the relative effects of anti-PD-1 on CD8+ T cells versus Treg cells may play an important role in dictating clinical outcomes127. Other PD-1hi T cells that can be preferentially impacted by PD-1 blockade and respond to this treatment are TFH and T follicular regulatory cells. These are the CD4+ T cells displaying the highest PD-1 expression; however, their dynamic changes and role in the response to immunotherapy are not fully understood. A population of circulating PD-1hiFoxp3–CD4+ T cells resembling TFH cells was described to be decreased during PD-1 blockade in patients with melanoma, but expanded upon anti-CTLA-4 (Fig. 2)61 according to observations in CTLA-4-deficient mice121,128. Intriguingly, these cells were found to suppress T cell effector functions in mice and humans and to accumulate in progressing melanoma tumors in mice61. Notably, in a retrospective analysis, patients with melanoma with lower frequencies of these TFH-like cells in PB showed improved responses to PD-1 blockade61. The precise role and predictive/prognostic significance of bona fide TFH cells in the response to immunotherapy need to be confirmed in prospective studies in patients.
Advances in sequencing technology are expected to tremendously accelerate the interrogation of these pharmacodynamic immune responses, through single-cell transcriptional characterization of T cell states129 and paired RNA–TCR sequencing analyses of clonally expanded T cells. Initial studies have shown that CD8+ TEX cells preferentially responding to ICB are more clonal than are other T cell states and are enriched for tumor-reactive cells across solid cancer types29,129,130. These initial results provide proof-of-principle that we will soon be able to monitor and interrogate the antigen-specific pharmacodynamic immune responses to immunotherapy at unparalleled depth.
Overcoming immunotherapy resistance by targeting melanoma metabolism
Despite the success of ICB, the efficacy of these therapies, even in combination, has reached a plateau, and novel classes of drugs are urgently needed. Tumor metabolic dependencies are emerging as key tumor vulnerabilies that may be amenable for targeting in combination with immunotherapy. Tumor cells generally adapt to undergo aerobic glycolysis, which is energetically less efficient than oxidative phosphorylation, but can feed metabolic intermediates into anabolic processes to sustain rapid cell division. Aerobic glycolysis is normally used by rapidly dividing cells, such as activated lymphocytes. However, in the TME, cancer cells are selected to acquire a metabolic advantage over normal immune cells, thus tipping the balance in favor of tumor progression and immune evasion.
Metabolic competition is particularly relevant in melanoma, as its oncogenic program unavoidably converges into activation of MAPK and PI3K–AKT signaling pathways—most commonly via activating BRAFV600E mutation or PTEN loss/inactivation—which supports a highly glycolytic profile131,132,133,134,135. Elevated glycolysis in human melanoma negatively correlates with T cell infiltration and activation122,134,136, and response to ACT or ICB134,137. Supporting these correlative observations in patients, pharmacologic or genetic targeting of glycolysis in tumor cells improves the activity of either ACT or ICB in mice122,134.
Intriguingly, progressing melanoma can acquire a hypermetabolic phenotype sustaining oxidative metabolism. These traits have been found to specifically distinguish brain melanoma metastases in humans138 and to contribute to ICB therapy resistance in mice139,140. To counteract this hypermetabolic phenotype and enhance immunotherapy, the antidiabetic biguanides are being investigated in melanoma, with promising results for phenformin in combination with anti-PD-1 in melanoma-bearing mice141. In addition, initial retrospective analyses show reduced occurrence of new brain metastases and trends toward favorable outcomes in individuals receiving metformin during ICB142.
Decreased oxygen tension in the microenvironment of hyper-oxidative tumors can promote T cell exhaustion143 and T cell resistance to anti-PD-1-mediated reinvigoration140 (Fig. 3). Conversely, tumor glycolysis and glucose deprivation in the microenvironment pose preferential resistance to CTLA-4 blockade122 (Fig. 3). The preferential barriers posed by oxidative versus glycolytic tumor metabolism to anti-PD-1 and anti-CTLA-4 blockade, respectively, may be at least partially explained by the distinct cellular localization of the direct targets of these immunotherapies. While PD-1 blockade primarily serves to reinvigorate dysfunctional PD-1+ T cells, which are pushed to terminal irreversible exhaustion states in low oxygen tension, CTLA-4 blockade has a role in counteracting Treg cells, whose stability is potentiated in glucose-deprived environments122,144 (Fig. 3). To counteract Treg function independent of tumor glycolysis, recent studies have shown that targeting lactate144 or fatty acid metabolism145 in Treg cells enhances the response to ICB in mouse melanoma models.

Schematic representation of possible metabolic scenarios in melanoma. Left, the pro-oncogenic program of melanoma, converging on MAPK and/or PI3K–AKT activation, supports a highly glycolytic profile, which results in a glucose-deprived and lactate-enriched environment that creates a metabolic barrier to glycolytic effector immune cells (for example NK cells and CTLs) and preferentially retains Treg cells and TEX cells due to their reliance on alternative sources of fuels (for example lactate and fatty acids, FA). This TME potentiates Treg stability and suppressive profiles and poses specific obstacles to the activity of CTLA-4 blockade. Right, the metabolic state of melanoma can evolve with tumor progression and metastasis dissemination through the acquisition of a hypermetabolic phenotype, including the ability to sustain oxidative metabolism, such as in brain metastases. Low oxygen tension fosters the development of terminal TEX cells, generating an environment that is particularly refractory to the activity of PD-1 blockade. OXPHOS, oxidative phosphorylation.
Together, these findings highlight three major concepts: (1) the existence of symbiotic metabolic interactions between tumors and dysfunctional/suppressive immune cells that use complementary sources of fuel in the TME; (2) the possibility to interfere with these vicious interactions as a useful strategy to combine with immunotherapy; and (3) the relevance of profiling tumor metabolic states as additional key information to guide therapeutic decisions for assigning patients to the right immunotherapy.
Conclusions and future directions
Despite its immunogenicity, metastatic melanoma grows and disseminates owing to immunosuppression and escape mechanisms. CTLA-4 and PD-1 blockade can efficiently target some of these mechanisms, by improving T cell priming, counteracting Treg suppression and re-invigorating PD-1hi TEX cells. However, many individuals still do not derive long-lasting benefits, even upon combined ICB.
In the era of combination immunotherapy, with more agents becoming available, the mechanism of single-agent therapies needs to be clearly delineated for guiding their rational combination. As the immunologic effects of immunotherapy occur early, we need to focus on these early events to identify (1) robust biomarkers, (2) resistance mechanisms, and (3) if necessary, proper therapies to add in a timely manner, in order to capitalize on the prior pharmacodynamic response. Moving forward, the neoadjuvant treatment setting appears to be an optimal platform to efficiently explore all these aspects. Moreover, toxicity from current and new immunotherapy combinations remains a critical point to address146. Understanding the molecular mediators of immune toxicity (for example, antibody mediated versus T cell mediated) will substantially help control these side effects and improve management of patients with melanoma.
The successful history of immunotherapy—especially with ICB—in melanoma has paved the way for its development in other cancers. Here, we have summarized the main lessons learned in the setting of melanoma immunobiology and immunotherapy, which can inform and accelerate further development of these treatments in other cancer types (Box 3). Comparing and contrasting efficacy and toxicity of immunotherapy across tumor types will tremendously aid in delineating the key parameters to assess for predicting response, limiting toxicity, and guiding therapeutic decisions to overcome treatment resistance.
References
Jakob, J. A. et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 118, 4014–4023 (2012).
Cohen, B. E., Manga, P., Lin, K. & Elbuluk, N. Vitiligo and melanoma-associated vitiligo: understanding their similarities and differences. Am. J. Clin. Dermatol. 21, 669–680 (2020).
Clark, W. H. Jr, From, L., Bernardino, E. A. & Mihm, M. C. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 29, 705–727 (1969).
Clark, W. H. Jr. et al. Model predicting survival in stage I melanoma based on tumor progression. J. Natl Cancer Inst. 81, 1893–1904 (1989).
Clemente, C. G. et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77, 1303–1310 (1996).
Azimi, F. et al. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 30, 2678–2683 (2012).
Thomas, N. E. et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J. Clin. Oncol. 31, 4252–4259 (2013).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526 (2011).
Pauken, K. E., Torchia, J. A., Chaudhri, A., Sharpe, A. H. & Freeman, G. J. Emerging concepts in PD-1 checkpoint biology. Semin. Immunol. 52, 101480 (2021).
Strome, S. E. et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 63, 6501–6505 (2003).
Iwai, Y. et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 20, 1239–1251 (2019).
Ascierto, P. A. et al. Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with nivolumab therapy: three-year follow-up of a randomized phase 3 trial. JAMA Oncol. 5, 187–194 (2019).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 372, 320–330 (2015).
Brahmer, J. R. et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010).
Wolchok, J. D. et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 40, 127–137 (2021)
Zappasodi, R., Wolchok, J. D. & Merghoub, T. Strategies for predicting response to checkpoint inhibitors. Curr. Hematol. Malig. Rep. 13, 383–395 (2018).
Ruscetti, F. W., Morgan, D. A. & Gallo, R. C. Functional and morphologic characterization of human T cells continuously grown in vitro. J. Immunol. 119, 131–138 (1977).
Kvistborg, P. et al. TIL therapy broadens the tumor-reactive CD8+ T cell compartment in melanoma patients. Oncoimmunology 1, 409–418 (2012).
Andersen, R. S. et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 72, 1642–1650 (2012).
Tran, E., Robbins, P. F. & Rosenberg, S. A. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat. Immunol. 18, 255–262 (2017).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Van den Eynde, B. J. & van der Bruggen, P. T cell defined tumor antigens. Curr. Opin. Immunol. 9, 684–693 (1997).
Lennerz, V. et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl Acad. Sci. USA 102, 16013–16018 (2005).
Oliveira, G. et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 596, 119–125 (2021).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Carreno, B. M. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Itzhaki, O. et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 34, 212–220 (2011).
Radvanyi, L. G. et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 18, 6758–6770 (2012).
Rosenberg, S. A. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011).
Goff, S. L. et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J. Clin. Oncol. 34, 2389–2397 (2016).
Linnemann, C. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81–85 (2015).
Lu, Y. C. et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J. Immunol. 190, 6034–6042 (2013).
Prickett, T. D. et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res. 4, 669–678 (2016).
Robbins, P. F. et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19, 747–752 (2013).
Tran, E. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015).
Krishna, S. et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370, 1328–1334 (2020).
Gilbert, A. E. et al. Monitoring the systemic human memory B cell compartment of melanoma patients for anti-tumor IgG antibodies. PLoS ONE 6, e19330 (2011).
Pfreundschuh, M. The genealogy of SEREX. Cancer Immun. 12, 7 (2012).
Sahin, U. et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc. Natl Acad. Sci. USA 92, 11810–11813 (1995).
Vijayasaradhi, S., Bouchard, B. & Houghton, A. N. The melanoma antigen gp75 is the human homologue of the mouse b (brown) locus gene product. J. Exp. Med. 171, 1375–1380 (1990).
Wang, R. F., Robbins, P. F., Kawakami, Y., Kang, X. Q. & Rosenberg, S. A. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumor-infiltrating lymphocytes. J. Exp. Med. 181, 799–804 (1995).
Livingston, P. O. et al. Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J. Clin. Oncol. 12, 1036–1044 (1994).
Cipponi, A. et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. 72, 3997–4007 (2012).
Ladanyi, A. et al. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother. 60, 1729–1738 (2011).
Sautes-Fridman, C., Petitprez, F., Calderaro, J. & Fridman, W. H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 19, 307–325 (2019).
Liu, W., Peng, Y. & Tobin, D. J. A new 12-gene diagnostic biomarker signature of melanoma revealed by integrated microarray analysis. PeerJ 1, e49 (2013).
Mortarini, R. et al. Constitutive expression and costimulatory function of LIGHT/TNFSF14 on human melanoma cells and melanoma-derived microvesicles. Cancer Res. 65, 3428–3436 (2005).
Erdag, G. et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 72, 1070–1080 (2012).
Garg, K. et al. Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum. Pathol. 54, 157–164 (2016).
Martinez-Rodriguez, M., Thompson, A. K. & Monteagudo, C. A significant percentage of CD20-positive TILs correlates with poor prognosis in patients with primary cutaneous malignant melanoma. Histopathology 65, 726–728 (2014).
Eschweiler, S. et al. Intratumoral follicular regulatory T cells curtail anti-PD-1 treatment efficacy. Nat. Immunol. 22, 1052–1063 (2021).
Joshi, N. S. et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity 43, 579–590 (2015).
Zlotnik, A., Burkhardt, A. M. & Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 11, 597–606 (2011).
Crawford, A. et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity 40, 289–302 (2014).
Zappasodi, R. et al. Non-conventional inhibitory CD4+Foxp3–PD-1hi T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell 33, 1017–1032 (2018).
Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Spranger, S. et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med. 5, 200ra116 (2013).
Taube, J. M. et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 4, 127ra137 (2012).
Zippelius, A. et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 64, 2865–2873 (2004).
Ahmadzadeh, M. et al. FOXP3 expression accurately defines the population of intratumoral regulatory T cells that selectively accumulate in metastatic melanoma lesions. Blood 112, 4953–4960 (2008).
Viguier, M. et al. Foxp3 expressing CD4+CD25high regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol. 173, 1444–1453 (2004).
Mukherji, B. et al. Clonal analysis of cytotoxic and regulatory T cell responses against human melanoma. J. Exp. Med. 169, 1961–1976 (1989).
Wang, H. Y., Peng, G., Guo, Z., Shevach, E. M. & Wang, R. F. Recognition of a new ARTC1 peptide ligand uniquely expressed in tumor cells by antigen-specific CD4+ regulatory T cells. J. Immunol. 174, 2661–2670 (2005).
Oliveira, G. et al. 655 Landscape of helper and regulatory CD4+ T cells in melanoma. J. Immunother. Cancer 9, A684 (2021).
Enninga, E. A., Nevala, W. K., Holtan, S. G., Leontovich, A. A. & Markovic, S. N. Galectin-9 modulates immunity by promoting Th2/M2 differentiation and impacts survival in patients with metastatic melanoma. Melanoma Res. 26, 429–441 (2016).
Lauerova, L. et al. Malignant melanoma associates with TH1/TH2 imbalance that coincides with disease progression and immunotherapy response. Neoplasma 49, 159–166 (2002).
Nevala, W. K. et al. Evidence of systemic TH2-driven chronic inflammation in patients with metastatic melanoma. Clin. Cancer Res. 15, 1931–1939 (2009).
Klarquist, J. S. & Janssen, E. M. Melanoma-infiltrating dendritic cells: limitations and opportunities of mouse models. Oncoimmunology 1, 1584–1593 (2012).
Marzagalli, M., Ebelt, N. D. & Manuel, E. R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol. 59, 236–250 (2019).
Pieniazek, M., Matkowski, R. & Donizy, P. Macrophages in skin melanoma-the key element in melanomagenesis. Oncol. Lett. 15, 5399–5404 (2018).
Tucci, M. et al. Immune system evasion as hallmark of melanoma progression: the role of dendritic cells. Front Oncol. 9, 1148 (2019).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015).
Spranger, S., Dai, D., Horton, B. & Gajewski, T. F. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723 e714 (2017).
Böttcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037 (2018).
Sun, X., Zhang, N., Yin, C., Zhu, B. & Li, X. Ultraviolet radiation and melanomagenesis: from mechanism to immunotherapy. Front Oncol. 10, 951 (2020).
Schwarz, A. et al. Langerhans cells are required for UVR-induced immunosuppression. J. Invest. Dermatol. 130, 1419–1427 (2010).
Wang, L., Jameson, S. C. & Hogquist, K. A. Epidermal Langerhans cells are not required for UV-induced immunosuppression. J. Immunol. 183, 5548–5553 (2009).
Nirschl, C. J. et al. IFNγ-dependent tissue-immune homeostasis is co-opted in the tumor microenvironment. Cell 170, 127–141.e115 (2017).
Khalil, D. N. et al. In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J. Clin. Invest. 129, 3435–3447 (2019).
Esensten, J. H., Helou, Y. A., Chopra, G., Weiss, A. & Bluestone, J. A. CD28 costimulation: from mechanism to therapy. Immunity 44, 973–988 (2016).
Blackburn, S. D. et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10, 29–37 (2009).
Huang, A. C. et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 25, 454–461 (2019).
Huang, A. C. et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017).
Kamphorst, A. O. et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl Acad. Sci. USA 114, 4993–4998 (2017).
Curiel, T. J. et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 9, 562–567 (2003).
Zappasodi, R., Merghoub, T. & Wolchok, J. D. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598 (2018).
Long, G. V. et al. Standard-dose pembrolizumab plus alternate-dose ipilimumab in advanced melanoma: KEYNOTE-029 cohort 1C, a phase 2 randomized study of two dosing schedules. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.Ccr-21-0793 (2021).
Maruhashi, T., Sugiura, D., Okazaki, I. M. & Okazaki, T. LAG-3: from molecular functions to clinical applications. J. Immunother. Cancer 8, e001014 (2020).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 386, 24–34 (2022).
Shen, R. et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 13, eabf5107 (2021).
Andrews, L. P. et al. Resistance to PD1 blockade in the absence of metalloprotease-mediated LAG3 shedding. Sci. Immunol. 5, eabc2728 (2020).
Eggermont, A. M. et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med. 375, 1845–1855 (2016).
Eggermont, A. M. M. et al. Adjuvant ipilimumab versus placebo after complete resection of stage III melanoma: long-term follow-up results of the European Organisation for Research and Treatment of Cancer 18071 double-blind phase 3 randomised trial. Eur. J. Cancer 119, 1–10 (2019).
Curti, B. D. & Faries, M. B. Recent advances in the treatment of melanoma. N. Engl. J. Med. 384, 2229–2240 (2021).
Eggermont, A. M. M. et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma (EORTC 1325-MG/KEYNOTE-054): distant metastasis-free survival results from a double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 22, 643–654 (2021).
Luke, J. J. Pembrolizumab versus placebo after complete resection of high-risk stage II melanoma: efficacy and safety results from the KEYNOTE-716 double-blind phase III trial. Ann. Oncol. 32, S1283–S1346 (2021).
Amaria, R. N. et al. Neoadjuvant systemic therapy in melanoma: recommendations of the International Neoadjuvant Melanoma Consortium. Lancet Oncol. 20, e378–e389 (2019).
Tarhini, A. A. et al. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 9, e87705 (2014).
Amaria, R. N. et al. Neoadjuvant and adjuvant nivolumab (nivo) with anti-LAG3 antibody relatlimab (rela) for patients (pts) with resectable clinical stage III melanoma. J. Clin. Oncol. 39, 9502–9502 (2021).
Menzies, A. M. et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the International Neoadjuvant Melanoma Consortium (INMC). Nat. Med. 27, 301–309 (2021).
Rawson, R. V. et al. Pathological response and tumour bed histopathological features correlate with survival following neoadjuvant immunotherapy in stage III melanoma. Ann. Oncol. 32, 766–777 (2021).
Rozeman, E. A. et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat. Med. 27, 256–263 (2021).
Blank, C. U. et al. First safety and efficacy results of PRADO: a phase II study of personalized response-driven surgery and adjuvant therapy after neoadjuvant ipilimumab (IPI) and nivolumab (NIVO) in resectable stage III melanoma. J. Clin. Oncol. 38, 10002–10002 (2020).
Moschos, S. J. et al. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J. Clin. Oncol. 24, 3164–3171 (2006).
Carthon, B. C. et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin. Cancer Res. 16, 2861–2871 (2010).
Ng Tang, D. et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol. Res. 1, 229–234 (2013).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Disco. 8, 1069–1086 (2018).
Arce Vargas, F. et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33, 649–663 (2018).
Romano, E. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145 (2015).
Sharma, A. et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. 25, 1233–1238 (2019).
Kavanagh, B. et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood 112, 1175–1183 (2008).
Selby, M. J. et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 1, 32–42 (2013).
Wing, J. B., Ise, W., Kurosaki, T. & Sakaguchi, S. Regulatory T cells control antigen-specific expansion of TFH cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 41, 1013–1025 (2014).
Zappasodi, R. et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 591, 652–658 (2021).
Korman, A. J. et al. Abstract SY09-01: next-generation anti-CTLA-4 antibodies. Cancer Res. 77, SY09-01 (2017).
Hashimoto, M. et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev. Med 69, 301–318 (2018).
Valpione, S. et al. Immune-awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat. Cancer 1, 210–221 (2020).
Kamada, T. et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl Acad. Sci. USA 116, 9999–10008 (2019).
Kumagai, S. et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 21, 1346–1358 (2020).
Sage, P. T., Paterson, A. M., Lovitch, S. B. & Sharpe, A. H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 41, 1026–1039 (2014).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020).
Li, H. et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789 (2019).
Falck Miniotis, M. et al. MEK1/2 inhibition decreases lactate in BRAF-driven human cancer cells. Cancer Res. 73, 4039 (2013).
Hall, A. et al. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the V600EBRAF oncogene. Oncotarget 4, 584–599 (2013).
Haq, R. et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 23, 302–315 (2013).
Cascone, T. et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 27, 977–987 e974 (2018).
Peng, W. et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 6, 202–216 (2016).
Brand, A. et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 24, 657–671 (2016).
Hugo, W. et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016).
Fischer, G. M. et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 9, 628–645 (2019).
Jaiswal, A. R. et al. Melanoma evolves complete immunotherapy resistance through the acquisition of a hypermetabolic phenotype. Cancer Immunol. Res. 8, 1365–1380 (2020).
Najjar, Y. G. et al. Tumor cell oxidative metabolism as a barrier to PD-1 blockade immunotherapy in melanoma. JCI Insight 4, e124989 (2019).
Kim, S. H. et al. Phenformin inhibits myeloid-derived suppressor cells and enhances the anti-tumor activity of PD-1 blockade in melanoma. J. Invest. Dermatol. 137, 1740–1748 (2017).
Afzal, M. Z., Mercado, R. R. & Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 6, 64 (2018).
Scharping, N. E. et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 22, 205–215 (2021).
Watson, M. J. et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651 (2021).
Wang, H. et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 21, 298–308 (2020).
Brahmer, J. R. et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 9, e002435 (2021).
Leko, V. & Rosenberg, S. A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 38, 454–472 (2020).
Coulie, P. G., Van den Eynde, B. J., van der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Turajlic, S. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 18, 1009–1021 (2017).
Acknowledgements
We would like to thank G. Linette, B. Carreno, R. Amaravadi, T. Mitchell, and J. D. Wolchok for their insightful feedback on this manuscript. A. C. H. is funded by grant K08 CA230157 from the NIH, the Doris Duke CSDA, Damon Runyon CIA, and funding from the Tara Miller Melanoma Foundation. R. Z. was supported by the Parker Institute for Cancer Immunotherapy Bridge Fellows Award. R. Z . acknowledges funding from the NCI SPORE (P50-CA192937).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
A. C. H. is a consultant for Immunai and receives research support from Bristol Myers Squibb. R. Z. is inventor on patent applications related to work on GITR, PD-1, and CTLA-4. R. Z. is a scientific advisory board member of iTEOS Therapeutics and receives grant support from AstraZeneca and Bristol Myers Squibb.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, A.C., Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: understanding the molecular basis for immune sensitivity and resistance. Nat Immunol 23, 660–670 (2022). https://doi.org/10.1038/s41590-022-01141-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-022-01141-1
This article is cited by
-
Targeting HDAC6 improves anti-CD47 immunotherapy
Journal of Experimental & Clinical Cancer Research (2024)
-
Augmenting MEK inhibitor efficacy in BRAF wild-type melanoma: synergistic effects of disulfiram combination therapy
Journal of Experimental & Clinical Cancer Research (2024)
-
Ultrasound-activated prodrug-loaded liposome for efficient cancer targeting therapy without chemotherapy-induced side effects
Journal of Nanobiotechnology (2024)
-
Mitofusin 1 silencing decreases the senescent associated secretory phenotype, promotes immune cell recruitment and delays melanoma tumor growth after chemotherapy
Scientific Reports (2024)
-
Inactivation of kindlin-3 increases human melanoma aggressiveness through the collagen-activated tyrosine kinase receptor DDR1
Oncogene (2024)
