
Abstract
A proportion of patients surviving acute coronavirus disease 2019 (COVID-19) infection develop post-acute COVID syndrome (long COVID (LC)) lasting longer than 12 weeks. Here, we studied individuals with LC compared to age- and gender-matched recovered individuals without LC, unexposed donors and individuals infected with other coronaviruses. Patients with LC had highly activated innate immune cells, lacked naive T and B cells and showed elevated expression of type I IFN (IFN-β) and type III IFN (IFN-λ1) that remained persistently high at 8 months after infection. Using a log-linear classification model, we defined an optimal set of analytes that had the strongest association with LC among the 28 analytes measured. Combinations of the inflammatory mediators IFN-β, PTX3, IFN-γ, IFN-λ2/3 and IL-6 associated with LC with 78.5–81.6% accuracy. This work defines immunological parameters associated with LC and suggests future opportunities for prevention and treatment.
Similar content being viewed by others


Main
Acute COVID-19, caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is characterized by a broad spectrum of clinical severity, from asymptomatic to fatal1,2. The immune response during acute illness contributes to both host defense and pathogenesis of severe COVID-19 (ref. 3). Pronounced immune dysregulation with lymphopenia and increased expression of inflammatory mediators3,4 have been described in the acute phase. Following acute COVID-19 infection, a proportion of patients develop physical and neuropsychiatric symptoms lasting longer than 12 weeks (known as Long COVID, chronic COVID syndrome or post-acute sequelae of COVID-19 (ref. 5)), henceforth denoted as LC. Although similar syndromes have been described following infection with SARS-CoV-1 (ref. 6) and Middle East respiratory syndrome–related coronavirus7, LC often develops after mild-to-moderate COVID-19 (refs. 8,9). Symptoms persisting 6 months were observed in 76% of hospitalized patients, with muscle weakness and fatigue being most frequently reported10,11. LC affects between 10% and 30% of community-managed COVID-19 cases 2 to 3 months after infection12,13 and can persist >8 months after infection14. LC symptoms include severe relapsing fatigue, dyspnea, chest tightness, cough, brain fog and headache15. The underlying pathophysiology of LC is poorly understood.
Here, we analyzed a cohort of individuals followed systematically for 8 months after COVID-19 infection according to a predefined schedule, comparing them to healthy donors unexposed to SARS-CoV-2 (unexposed healthy controls (UHCs)) before December 2019, and individuals who had been infected with prevalent human coronaviruses (HCoVs; HCoV-NL63, O229E, OC43 or HKU1), but not SARS-CoV-2. The ADAPT study9 enrolled adults with SARS-CoV-2 infections confirmed by PCR at St Vincent’s Hospital community-based testing clinics in Sydney (Australia). For the majority of participants, their first visit occurred between months 2 and 3 after infection (median of 79 days after the date of initial diagnosis)9,14, with 93.6% and 84.5% of participants completing subsequent month 4 (median, 128 days) and month 8 (median, 232 days) visits (Table 1). Of the 147 patients recruited (70.5% through ADAPT sites and 29.5% externally), 31 participants (21.08%) were designated as LC based on the occurrence of one of three major symptoms (fatigue, dyspnea or chest pain) at month 4 (Supplementary Table 1). These participants were age and gender matched with 31 asymptomatic matched controls (MCs) from the same cohort who did not report symptoms at month 4 after infection but were symptomatic during the acute phase of the infection (Supplementary Table 2). There was a 10% trend toward some improvement of symptoms over time in LC, but this trend was not statistically significant (Fisher’s exact P = 0.44).
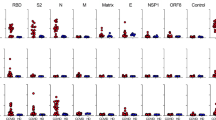
To examine biomarkers associated with LC, we assessed 28 analytes in the serum of patients from the LC, MC, HCoV and UHC groups at month 4 after infection using a bead-based assay. Six proinflammatory cytokines (interferon β (IFN-β), IFN-λ1, IFN-γ, CXCL9, CXCL10, interleukin-8 (IL-8) and soluble T cell immunoglobulin mucin domain 3 (sTIM-3)) were elevated in the LC and MC groups compared to the HCoV and UHC groups (Fig. 1), with no difference observed in the 22 other analytes, including IL-6 and IL-33 (Extended Data Fig. 1). There was no difference between LC and MCs for any individual analyte at this time point (Extended Data Fig. 1a, b). IFN-β was 7.92-fold and 7.39-fold higher in the LC and MC groups compared to the HCoV group and 7.32- and 6.83-fold higher compared to UHCs (Fig. 1a). IFN-λ1 was increased 2.44-fold and 3.24-fold in the LC and MC groups compared to the HCoV group and 2.42- and 3.21-fold compared to UHCs. IL-8 was higher in the LC (3.43-fold) and MC (3.56-fold) groups compared to the HCoV and UHC groups (Fig. 1a). CXCL10 was elevated in the LC group compared to the HCoV (2.15-fold) and UHC (3.2-fold) groups and in the MC group compared to the HCoV (1.7-fold) and UHC (3.06-fold) groups. CXCL9 was 1.69-fold higher in the LC group than in the UHC group, and sTIM-3 was elevated in the LC group, but not the MC group, when compared to the HCoV group (1.46-fold) (Fig. 1a and Extended Data Fig. 1c).

a, Higher levels of IFN-β, IFN-λ1, CXCL9, CXCL10, IL-8 and sTIM-3 at 4 months in the LC group (n = 31) and MC (n = 31) compared to individuals infected with common cold coronavirus (HCoV) (n = 25) and unexposed healthy donors (n = 13). b, Reduction of cytokine levels at 8 months. IFN-β and IFN-λ1 levels were higher in the LC group than the MC group at month 8. c, ACE2 activity increased at months 3 and 4 after SARS-CoV-2 infection and then decreased at 8 months in both groups. ACE2 activity in the HCoV and unexposed healthy donors (UHC) groups was minimal. Data are shown as medians with error bars representing interquartile ranges (IQRs). Kruskal–Wallis with Dunn’s correction was used for the 4-month analysis; two-tailed P values <0.05 (*), <0.01 (**), <0.001 (***) and <0.0001 (****) were considered significant. A Wilcoxon t test was used for paired comparison of 4- and 8-month data, and a Mann–Whitney t test was used for unpaired samples. HCoV, human endemic coronavirus infected; M, months; NS, not significant.
IFN-β and IFN-λ1 decreased 4.4-fold and 1.8-fold, respectively, in the MC group at month 8 compared to month 4 (Fig. 1b). In the LC group, IFN-β decreased by 1.5-fold, and IFN-λ1 increased by 1.05-fold at month 8 compared to month 4, which was not statistically significant (Fig. 1b). At month 8, IFN-β and IFN-λ1 remained significantly elevated in the LC group compared to the MC, HCoV and UHC groups (Extended Data Fig. 2a). Reductions in CXCL9, CXCL10, IL-8 and sTIM-3 were observed in the LC and MC groups at month 8 compared to month 4 (Fig. 1b). At month 8, there was also decreased expression of some of the 22 analytes that were not significantly different among the four groups at month 4 (Extended Data Fig. 2b,c).
Because plasma ACE2 activity has been reported to be elevated 114 days after SARS-CoV-2 infection16, we investigated whether this occurred in our cohort at months 3, 4 and 8 after infection. Median plasma ACE2 activity was significantly higher in both LC and MC groups compared to the HCoV group at month 3 (LC, 1.92-fold; MC, 2.47-fold) and month 4 (LC, 1.75-fold; MC, 2.62-fold) after infection (Fig. 1c). At month 8, plasma ACE2 activity in the LC and MC groups decreased to levels observed in the HCoV and UHC groups (Fig. 1c). No difference was observed within LC and MC groups at months 3, 4 or 8, but both groups had higher activity compared to the HCoV group, suggesting that this parameter is specific to SARS-CoV-2 infection and is not a common feature of other coronaviruses.
Next, we used a classification model to determine an optimal set of analytes most strongly associated with LC. This linear classifier was trained on log-transformed analyte data to reduce the bias observed in each of the analytes and improve model accuracy. This log-linear classification model was used to develop a metric for feature importance17. To identify analytes that were associated with LC and not MC, we used the analyte data at month 8, the time point with the greatest difference between the LC and MC groups. The performance of each of the log-linear models was quantified by an accuracy estimate and an F1 score evaluated by taking averages after bootstrapping, which randomly sampled from the original population to create a new population. By considering every possible pair of the 28 serum analytes and plasma ACE2 activity, a classification model including two analytes (IFN-β and pentraxin 3 (PTX3)) had an LC prognostic accuracy of 78.54% and an F1 score of 0.77. Three analytes (IFN-β, PTX3 and IFN-γ) achieved an accuracy of 79.68%, with an F1 score of 0.79. Four analytes (IFN-β, PTX3, IFN-λ2/3 and IL-6) achieved an accuracy of 81.59% and an F1 score of 0.81. When all 29 analytes were featured, the calculated accuracy was 77.4%, with an F1 score of 0.76 (Table 2).
After generating 1,000 randomly sampled populations, we counted the number of times each feature appeared in the best performing set of features, combining sets if several sets achieved the same accuracy. This revealed that IFN-β was the most important feature, appearing in 89%, 93% and 94% of the best sets of two, three and four features, respectively (Fig. 2a). Linear classifiers defined a decision boundary. Each patient analyte concentration at month 8 lied on either side of the boundary, and its positioning relative to the boundary determined whether the patient was predicted to experience LC or asymptomatic COVID (Fig. 2b). Although the decision boundary of the four featured analytes at month 8 is four dimensional, the boundary can be visualized with two-dimensional projections of IFN-β against the other highly associated analytes (PTX3, IFN-γ, IFN-λ2/3 and IL-6 (Fig. 2b). Longitudinal levels of these key feature cytokines indicate the advantage of log-linear models in differentiating LC from MCs (Fig. 2c).

a, Representative bar graph of long-linear classification data showing the frequency of analytes appearing as the top features highly associated with LC following 1,000 sampled populations. b, Two-dimensional scatterplot separating LC (red dots) and MCs (blue dots) with a decision boundary (straight light) based on the concentrations of IFN-β and PTX3, IFN-β versus IFN-𝛾, IFN-β versus IFN-λ2/3 and IFN-β versus IL-6. Data points found above the decision boundary have a strong association with LC. c, Representation of the longitudinal levels of the five key cytokines. Data are shown as medians with error bars representing interquartile ranges (IQRs). Two-tailed P values <0.01 (**), <0.001 (***) and <0.0001 (****) were considered significant. A Wilcoxon t test was used for paired samples, and a Mann–Whitney t test was used for unpaired samples.
To investigate differences in immune cell profiles between LC and MCs, we developed a 19-parameter flow cytometry panel and phenotyped peripheral blood mononuclear cells (PBMCs) from LC and MC donors at months 3 and 8 after infection. Dimensional reduction via TriMap coupled with Phenograph clustering (n = 14; LC = 7, MC = 7) identified 24 distinct cell clusters at month 3 and 21 clusters at month 8 (Extended Data Fig. 3a) including T, B, NK and myeloid cell clusters (Extended Data Fig. 3b,c). Concatenated phenotype data from each of the 7 LC or MC and 7 UHC contributed to every population cluster (Extended Data Fig. 4a–d). Of the 24 subsets identified at month 3, five were absent in LC donors: naive CD127lowGzmB−CCR7+CD45RA+CD27+CD8+ T cells, CD57+GPR56+GzmB+CD8+ T cells, naive CD127loTIM-3−CCR7+CD45RA+CD27+CD4+ T cells, innate-like CD3+CD4−CD8− T cells (may comprise natural killer T cells and γδ-T cells), and naive CD127loTIM-3−CD38lowCD27−IgD+ B cells (Fig. 3a). Three clusters remained absent at month 8 in LC donors (naive CD127lowGzmB−CCR7+CD45RA+CD27+CD8+ T cells, naive CD127lowTIM-3−CCR7+CD45 RA+CD27+CD4+ T cells, and naive CD127lowTIM-3−CD38lowCD27−IgD+ B cells) (Fig. 3b), indicating perturbations at month 8 in LC donors. Naive T and B cells expressing low levels of CD127 and TIM-3 were detected in the MC and UHC groups but were absent in the LC group at months 3 and 8 (Extended Data Fig. 4e,f).

a, Dimensional reduction utilizing TriMap and clustering with phenograph was used to visualize immune cell phenotypes. Five populations consisting of unactivated naive and cytotoxic phenotypes were absent in the LC group when compared to MCs at 3 months (n = 14, seven samples per group). Absent populations are outlined in red (middle and right panel), with median percentages of the LC and MC contribution to each cluster population shown in the bar graph. b, Unactivated naive T and B cell populations with low expression of CD127 and no TIM-3 expression remained absent in peripheral blood at 8 months in the LC group (clusters 1, 3 and 5). The phenotypes of the clusters are as follows: (1) CD127lowGzmB−CCR7+CD45RA+CD27+ naive CD8+ T cells, (2) CD57+ highly cytotoxic (GPR56+GzmB+) CD8+ T cells, (3) CD127lowTIM-3−CCR7+CD45RA+CD27+ naive CD4+ T cells, (4) CD3+CD4−CD8− innate-like T cells (may comprise natural killer T cells and γδ-T cells) and (5) naïve CD127lowTIM-3−CD38lowCD27−IgD+ B cells. c, Activated nonlymphoid (myeloid) cells with combined expression of CD38 and HLA-DR in the LC and MC groups at 3 months; levels were not significantly reduced at 8 months in the LC group. Elevated levels of activated monocytes and pDCs were found in the LC group when compared to MCs at 3 and 8 months. d, No difference in PD-1 levels was found on CD4+ T cells, but higher expression by CD8+ T cells was found in the LC group at both time points. Higher TIM-3 expression on CD8+ T cells was observed in the LC group at 3 months. Data are shown as median with IQR. Two-tailed P values <0.05 (*), <0.01 (**), <0.001 (***) and <0.0001 (****) were considered significant. A Wilcoxon t test was used for paired samples, and a Mann–Whitney t test was used for unpaired samples.
The frequency of highly activated CD38+HLA-DR+ myeloid cells was elevated at month 8 in the LC group compared to MCs (Fig. 3c). Frequencies of activated CD14+CD16+ monocytes were higher in the LC group compared to MCs at months 3 and 8. The percentages of plasmacytoid dendritic cells (pDCs) expressing the activation markers CD86 and CD38 were also higher in the LC group at both time points compared to MCs (Fig. 3c). There was no difference in the frequencies of activated CD11c+ myeloid dendritic cells between month 3 and month 8 (Extended Data Fig. 5a). The T cell activation and exhaustion markers PD-1 and TIM-3 were more highly expressed on CD8+ T cells in the LC group compared to MCs at month 3 (PD-1, 3.04-fold; TIM-3, 1.6-fold) and month 8 (PD-1 2.86-fold) (Fig. 3d). However, PD-1 and TIM-3 coexpression was similar on CD4+ and CD8+ T cells in the LC and MC groups (Extended Data Fig. 5b).
Here, we show that convalescent immune profiles after COVID-19 are different from those following infection with other coronaviruses. Several cytokines (mostly type I and III IFN, but also chemokines downstream of IFN-γ) were highly elevated in individuals following the resolution of active SARS-CoV-2 infection compared to HCoVs and UHCs at month 4 after infection. IFN-β and IFN-λ1 remained elevated in the LC group at month 8 after initial infection, while their levels began to resolve in MCs. Elevated plasma ACE2 activity was noted in the LC and MC groups at month 4 but trended toward normal by month 8 after infection. We identified a set of analytes (IFN-β, PTX3, IFN-γ, IFN-λ2/3 and IL-6) that highly associated with LC at month 8, indicating that components of the acute inflammatory response and activation of fibroblast or epithelial cells, T cells and myeloid cells are associated with LC. Immune cell phenotyping indicated chronic activation of a subset of CD8+ T cells, with expansion of PD-1+ and TIM-3+ subsets and pDCs and monocytes persisting from month 3 to month 8 in the LC group. These changes were accompanied by an absence of naive T and B cell subsets expressing low levels of CD127 and TIM-3 in peripheral blood of patients with LC. These findings suggest that SARS-CoV-2 infection exerts unique prolonged residual effects on the innate and adaptive immune systems and that this may be driving the symptomology known as LC.
IFN-β and IFN-λ1 were highly elevated in convalescent COVID-19 samples compared to HCoV and UHC samples. Although these levels decreased over time in patients who recovered, they remained high in patients with LC. The morbidity of acute COVID-19 infection appears to correlate with high expression of type I and III IFN in the lungs of patients18. IFN-λ produced by murine lung dendritic cells in response to synthetic viral RNA is associated with damage to lung epithelium19, and IFN-λ signaling hampers lung repair during influenza infection in mice20. Severe acute COVID-19 has been associated with diminished type I IFN and enhanced IL-6 and tumor necrosis factor (TNF) responses19. Although our cohort of individuals with LC consisted mostly of patients with mild or moderate initial illness, elevated type I and III IFN levels were maintained to month 8 after infection and are consistent with the observed prolonged activation of pDCs, indicating a chronic inflammatory response.
Patients with COVID-19 who are admitted to the intensive care unit have high plasma levels of sTIM-3 (ref. 21). We found elevated levels of sTIM-3 in the LC group, but not in the MC or HCoV groups, which is consistent with the expanded subsets of memory CD8+ T cells expressing TIM-3 and PD-1 and indicates chronic T cell activation and potentially exhaustion. Similarly, shedding of membrane-bound protein ACE-2 during acute infection22 resulting in increased activity in plasma16 continues into convalescence, regardless of symptom severity at month 4, and normalizes at month 8 in most patients.
We employed a log-linear classification model to assess all combinations of analytes to determine the subset of analytes most strongly associated with LC. IFN-β, together with PTX3, IFN-λ2/3, IFN-γ and IL-6, differentiated LC from MCs with high accuracy at month 8. IFN-λ2/3 are secreted by pDCs following viral RNA sensing by TLR7, TLR9 and RIG-123,24. PTX3 increased in lung epithelia and plasma of patients with severe COVID-19 and can serve as an independent strong prognostic indicator of short-term mortality25,26,27. IL-6 is a pleiotropic mediator that drives inflammation and immune activation28. A high IL-6/IFN-γ ratio is associated with severe acute COVID-19 infection29. The observation that the best correlate for LC is an eclectic combination of biomarkers reinforces the breadth of host response pathways that are activated during LC.
T cell activation (indicated by CD38 and HLA-DR), T cell exhaustion and increases in B cell plasmablasts occur during severe COVID-19 (refs. 30,31,32). These markers identified highly activated monocytes and pDCs, the frequencies of which decreased over time in MCs, but not in patients with LC. Type I and type III IFN upregulate major histocompatibility complex expression, including HLA-DR33. An unbiased large-scale dimensional reduction approach identified the depletion of three clusters of naive B and T cell subsets present in the LC group at month 8 after infection. Altogether, these observations suggest persistent conversion of naive T cells into activated states, potentially due to bystander activation secondary to underlying inflammation and/or antigen presentation by activated pDCs or monocytes. The ultimate result of this chronic stimulation may be expansion of PD-1+ or TIM-3+ CD8+ memory T cells. Bystander activation of unactivated naive subsets into more activated phenotypes is consistent with observations in acute severe COVID-19 (refs. 34,35).
Although individuals with LC and MCs were matched for age and gender, it is possible that the differences observed reflect differences in unrecognized factors between these groups. Although more LC donors had severe acute disease (eight LC donors and two MCs), sensitivity analyses excluding these patients did not alter the statistical significance of the major associations described here. Because of the timing of ethics approval and cohort setup, samples were not collected during acute infection. We were therefore unable to determine whether elevations in biomarkers during convalescence correlate with levels during acute infection. Although some perturbations observed here are potentially consistent with a hypothesis that the major drivers of the expression of biomarkers in convalescence are those in the acute infection, others are not. Our results require validation in other LC cohorts. Finally, our definition of LC was set internally given the lack of international consensus. Nevertheless, the inclusion of three of the most common persisting symptoms and blinding of cases and controls helped ensure the validity of our findings.
In summary, our data indicate an ongoing, sustained inflammatory response following even mild-to-moderate acute COVID-19, which is not found following prevalent coronavirus infection. The drivers of this activation require further investigation, but possibilities include persistence of antigen, autoimmunity driven by antigenic cross-reactivity or a reflection of damage repair. These observations describe an abnormal immune profile in patients with COVID-19 at extended time points after infection and provide clear support for the existence of a syndrome of LC. Our observations provide an important foundation for understanding the pathophysiology of this syndrome and potential therapeutic avenues for intervention.
Methods
Cohort characteristics
The ADAPT study is a prospective cohort study of post–COVID-19 recovery established in April 2020 (ref. 14). A total of 147 participants with confirmed SARS-CoV-2 infection were enrolled, the majority following testing in community-based clinics run by St Vincent’s Hospital Sydney, with some patients also enrolled with confirmed infection at external sites. Initial study follow-up was planned for 12 months after COVID-19 and subsequently extended to 2 years. Extensive clinical data and a biorepository was systematically collected prospectively. The aims of ADAPT are to evaluate a number of outcomes after COVID-19 relating to pathophysiology, immunology and clinical sequalae. Laboratory testing for SARS-CoV-2 was performed using nucleic acid detection from respiratory specimens with the EasyScreen Respiratory Detection kit (Genetic Signatures) and the EasyScreen SARS-CoV-2 detection kit. Two ADAPT cohort subpopulations were defined based on initial severity of COVID-19 illness: (1) patients managed in the community and (2) patients admitted to the hospital for acute infection (including those requiring intensive care support for acute respiratory distress syndrome). Patients were defined as having LC at 4 months based on the presence of one or more of the following symptoms: fatigue, dyspnea or chest pain14. These patients were gender and age (±10 years) matched with ADAPT participants without LC (matched ADAPT controls) (Table 1). Samples for these analyses were collected at the 3-, 4- and 8-month assessments. Our cohort consisted of 62 participants (31 with LC and 31 MCs); enrollment visits were performed at a median of 76 (IQR, 64–93) days after initial infection. Their 4-month assessments were performed at a median of 128 (IQR, 115–142) days after initial infection (4.2 months). Their 8-month assessments were performed at a median of 232 (IQR, 226–253) days after initial infection (7.7 months). The dropout rate has been very low to date (approximately 9.4% at 12 months). Four participants did not complete the 8-month assessment after the 4-month assessment. The reasons for this include ‘did not attend’ (n = 2) and ‘lost to follow-up’ (n = 2). A further population of patients presenting to St Vincent’s Hospital clinics for COVID-19 testing on the multiplex respiratory panel who were PCR positive for any of the four human common cold coronaviruses (HCoV-NL63, O229E, OC43 or HKU1) and PCR negative for SARS-CoV-2 were recruited into the ADAPT-C substudy and used as a comparator group.
Ethics
The ADAPT study was approved by the St Vincent’s Hospital Research Ethics Committee (2020/ETH00964) and is a registered trial (ACTRN12620000554965). The ADAPT-C substudy was approved by the same committee (2020/ETH01429). All data were stored using REDCap (v11.0.3) electronic data capture tools. Unexposed healthy donors were recruited through St Vincent’s Hospital and approved by St Vincent’s Hospital Research Ethics Committee (HREC/13/SVH/145). The University of Melbourne unexposed donors were approved by Medicine and Dentistry HESC study ID 2056689. All participants gave written informed consent, and patients were not compensated.
Sample processing and flow cytometry
Blood was collected for biomarker analysis (serum separating tube (SST) 8.5 ml x1 (serum) and EDTA 10 ml x1 (plasma)), and 36 ml was collected for PBMCs (ACD (citric acid, trisodium citrate and dextrose) 9 ml x4). Phenotyping of PBMCs was performed as described previously36. Briefly, cryopreserved PBMCs were thawed using RPMI (+L-glutamine) medium (ThermoFisher Scientific) supplemented with penicillin/streptomycin (Sigma-Aldrich) and subsequently stained with antibodies binding to extracellular markers for 20 min. Extracellular panel included Live/Dead dye Near InfraRed, CXCR5 (MU5UBEE) and CD38 (HIT2) (ThermoFisher Scientific); CD3 (UCHT1), CD8 (HIL-72021), PD-1 (EH12.1), TIM-3 (TD3), CD27 (L128), CD45RA (HI100), CD86 (BU63), CD14 (HCD14), CD16 (GB11), IgD (IA6-2), CD25 (2A3) and CD19 (HIB19) (BioLegend); and CD4 (OKT4), CD127 (A019D5), HLA-DR (L234), GRP56 (191B8), CCR7 (G043H7) and CD57 (QA17A04) (BD Biosciences). Perm Buffer II (BD Pharmingen) was used for intracellular staining of granzyme B (GB11, BD Biosciences). Samples were acquired on an Cytek Aurora (BioLegend) using Spectroflo software. Before each run, all samples were fixed in 0.5% paraformaldehyde.
Serum analytes
The LEGENDplex Human Anti-Virus Response Panel (IL-1β, IL-6, IL-8, IL-10, IL-12p70, IFN-α2, IFN-β, IFN-λ1, IFN-λ2/3, IFN-γ, TNF-α, IP-10 and GM-CSF) and a custom-made panel (IL-5, IL-9, IL-13, IL-33, PD-1, sTIM-3, sCD25, CCL2 (MCP-1), PTX3, transforming growth factor β1, CXCL9 (MIG-1), myeloperoxidase, PECAM-1, ICAM-1 and VCAM-1) were purchased from BioLegend, and assays were performed as per the manufacturer’s instructions. Beads were acquired and analyzed on a BD Fortessa X20 SORP (BD Biosciences). Samples were run in duplicate, and 4,000 beads were acquired per sample. Data analysis was performed using Qognit LEGENDplex software (BioLegend). Lower limit of detection values were used for all analytes at the lower limit.
Catalytic ACE2 detection in plasma
Plasma ACE2 activity was measured using a validated, sensitive quenched fluorescent substrate-based assay as previously described37. Briefly, plasma (0.25 ml) was diluted into low-ionic-strength buffer (20 mmol l−1 Tris-HCl, pH 6.5) and added to 200 ml ANXSepharose 4 Fast-Flow resin (Amersham Biosciences, GE Healthcare) that removed a previously characterized endogenous inhibitor of ACE2 activity. After binding and washing, the resulting eluate was assayed for ACE2 catalytic activity. Duplicate samples were incubated with the ACE2-specific quenched fluorescent substrate, with or without 100 mM ethylenediaminetetraacetic acid. The rate of substrate cleavage was determined by comparison to a standard curve of the free fluorophore 4-amino-methoxycoumarin (Sigma-Aldrich) and expressed as picomoles of substrate cleaved per milliliter of plasma per minute. The intra- and interassay coefficients of variation were 5.6% and 11.8%, respectively. Samples below the limit of detection were designated 0.02 (half the lower limit of detection; i.e., 50% × 0.04).
Linear model
The analytes most associated with LC were identified via log-linear classification. For an arbitrary set of four analytes, let the concentration of the ith analyte at 8 months be denoted wi. Log-linear classification assigns a weight ai to the logarithm of each analyte concentration. A linear function of these logged concentrations and weights takes the form \(f\left( {\vec a} \right)\) is a threshold parameter. The weights wi and the intercept w0 are selected to maximize the predictive power of the linear classifier by training on the analyte data, where \(f\left( {\vec a} \right) > 0\) results in the classifier predicting that the participant with analyte concentration \(\vec a\) has LC and does not have LC otherwise.
Because of the modest small sample size of 58 participants at month 8, we performed bootstrapping to randomly sample new populations of size 58 from our population with replacement. The sampled population was then split 29:29 into test and train datasets. The training dataset was used to train a log-linear classifier using Python3 v3.8.10 and the Scikit-learn machine learning package v0.24.1. From the test set, the number of true positives (TPs; both the classifier and data indicate the participant had LC), true negatives (TNs; both the classifier and data indicate the participant had asymptomatic COVID), false positives (FPs; classifier predicts the participant will have LC, but the data disagree) and false negatives (FNs; classifier predicts the participant will have asymptomatic COVID, but the data disagree) were identified. Then, two subsequent scores were calculated. The accuracy is defined as (TP + TN)/(TP + TN + FP + FN) and measures the proportion of test participants that had their COVID status correctly predicted. The second measure is the F1 score and is defined as TP/(TP + 0.5 × (FP + FN)), which is a measure that combines recall, how many LC cases were correctly predicted and precision (of all the participants predicted to have LC, how many were correct). This process is repeated for 1,000 different bootstrapped sample populations. The average accuracy of a model of N analytes is then calculated and used to assess which combination of N analytes performs the best.
Dimensional reduction and clustering analysis
FCS 3.0 files were compensated manually using acquisition-defined matrix as a guide, and the gating strategy was based on unstained or endogenous controls. Live singlets were gated from patients with LC and asymptomatic MCs using FlowJo v.10.7.2, samples were decoded and statistical analysis between groups and unsupervised analysis was performed, with matched asymptomatic controls as the primary comparator group. For unsupervised analysis, the following FlowJo plugins were used: DownSample (v.3), TriMap (v.0.2), Phenograph (v.3.0) and ClusterExplorer (v.1.5.9) (all FlowJo LLC). First, 100,000 events per sample were downsampled from the total live singlet gate (Extended Data Fig. 6). The newly generated FCS files were labeled according to control or patient group (LC or MCs) and concatenated per group. Subsequently, 20,000 events were taken from each grouped sample by downsampling. The two new FCS files corresponding to LC and MCs were then concatenated for dimensionality reduction analysis using TriMap (40,000 events in total). TriMap was conducted using the following parameters to include the markers CD25, CD38, CCR7, CD19, IgD, CD45RA, PD-1, TIM-3, CD4, CD57, CD127, CD27, HLA-DR, CD8, CXCR5, GPR56 and granzyme B and using the following conditions: metric = Euclidean, nearest neighbors = 15 and minimum distance = 0.5. The phenograph plugin was then used to determine clusters of phenotypically related cells. The same markers as TriMap and parameters k = 152 and Run ID = auto were used for analysis. Finally, the ClusterExplorer plugin was used to identify the phenotype of the clusters generated by phenograph.
Statistical analysis
All column graphs are presented as medians with IQRs. One-way analysis of variance with Kruskal–Wallis and Dunn’s correction for multiple comparisons was used for serum analyte analysis. A Wilcoxon paired t test was used to analyze statistical data with Prism v9.0 (GraphPad) software. For unpaired samples, a Mann–Whitney U test was used. Two-tailed P values less than 0.05 were considered significant (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
To protect patient privacy, underlying electronic health records may be accessed via a remote server pending a material transfer agreement and approval from study steering committee. As data within this manuscript are from an ongoing clinical trial, further data will be provide by the corresponding author upon request and will require approval from study steering committee.
References
Sudre, C. H. et al. Attributes and predictors of long COVID. Nat. Med. 27, 626–631 (2021).
Wiersinga, W. J., Rhodes, A., Cheng, A. C., Peacock, S. J. & Prescott, H. C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA 324, 782–793 (2020).
Carvalho, T., Krammer, F. & Iwasaki, A. The first 12 months of COVID-19: a timeline of immunological insights. Nat. Rev. Immunol. 21, 245–256 (2021).
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Peluso, M. J., et al. Rapid implementation of a cohort for the study of post-acute sequelae of SARS-CoV-2 infection/COVID-19. Preprint at medRxiv https://doi.org/10.1101/2021.03.11.21252311 (2021).
Lam, M. H. et al. Mental morbidities and chronic fatigue in severe acute respiratory syndrome survivors: long-term follow-up. Arch. Intern. Med. 169, 2142–2147 (2009).
Ahmed, H. et al. Long-term clinical outcomes in survivors of severe acute respiratory syndrome and Middle East respiratory syndrome coronavirus outbreaks after hospitalisation or ICU admission: a systematic review and meta-analysis. J. Rehabil. Med 52, jrm00063 (2020).
Halpin, S. J. et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: a cross-sectional evaluation. J. Med Virol. 93, 1013–1022 (2021).
Darley, D. R. et al. Persistent symptoms up to four months after community and hospital-managed SARS-CoV-2 infection. Med J. Aust. 214, 279–280 (2021).
Cortinovis, M., Perico, N. & Remuzzi, G. Long-term follow-up of recovered patients with COVID-19. Lancet 397, 173–175 (2021).
Huang, C. et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 397, 220–232 (2021).
Sudre, C. H., et al. Anosmia and other SARS-CoV-2 positive test-associated symptoms, across three national, digital surveillance platforms as the COVID-19 pandemic and response unfolded: an observation study. Preprint at medRxiv https://doi.org/10.1101/2020.12.15.20248096 (2020).
Tenforde, M. W. et al. Characteristics of adult outpatients and inpatients with COVID-19 - 11 academic medical centers, United States, March-May 2020. MMWR Morb. Mortal. Wkly Rep. 69, 841–846 (2020).
Darley, D. et al. Limited recovery from post-acute sequelae of SARS-CoV-2 (PASC) at eight months in a prospective cohort. ERJ Open Res. 7, 00384–2021 (2021).
Lopez-Leon, S. et al. More than 50 long-term effects of COVID-19: a systematic review and meta-analysis.Sci. Rep. 11, 16144 (2021).
Patel, S. K. Plasma ACE2 activity is persistently elevated following SARS-CoV-2 infection: implications for COVID-19 pathogenesis and consequences.Eur. Respir. J. 57, 2003730 (2021).
Dunn, P. K. Log-linear models. In Encyclopedia of Social Measurement (Ed. Kempf-Leonard, K.) 585–589 (Elsevier, 2005).
Broggi, A. et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369, 706–712 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Major, J. et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 712–717 (2020).
Ueland, T. et al. Elevated plasma sTIM-3 levels in patients with severe COVID-19. J. Allergy Clin. Immunol. 147, 92–98 (2021).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Portela Sousa, C. & Brites, C. Immune response in SARS-CoV-2 infection: the role of interferons type I and type III. Braz. J. Infect. Dis. 24, 428–433 (2020).
Kelly, A. et al. Immune cell profiling of IFN-lambda response shows pDCs express highest level of IFN-lambdaR1 and are directly responsive via the JAK-STAT pathway. J. Interferon Cytokine Res. 36, 671–680 (2016).
Norata, G. D. et al. Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation 120, 699–708 (2009).
Kimura, S. et al. Relationships of elevated systemic pentraxin-3 levels with high-risk coronary plaque components and impaired myocardial perfusion after percutaneous coronary intervention in patients with ST-elevation acute myocardial infarction. Circ. J. 78, 159–169 (2014).
Brunetta, E. et al. Macrophage expression and prognostic significance of the long pentraxin PTX3 in COVID-19. Nat. Immunol. 22, 19–24 (2021).
Tanaka, T., Narazaki, M. & Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 6, a016295 (2014).
Lagunas-Rangel, F. A. & Chavez-Valencia, V. High IL-6/IFN-gamma ratio could be associated with severe disease in COVID-19 patients. J. Med Virol. 92, 1789–1790 (2020).
Wang, Z. et al. COVID-19 severity correlates with weaker T-cell immunity, hypercytokinemia, and lung epithelium injury. Am. J. Respir. Crit. Care Med 202, 606–610 (2020).
Mathew, D. et al. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. Science 369, 6508 (2020).
Koutsakos, M. et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Rep. Med 2, 100208 (2021).
Keskinen, P., Ronni, T., Matikainen, S., Lehtonen, A. & Julkunen, I. Regulation of HLA class I and II expression by interferons and influenza A virus in human peripheral blood mononuclear cells. Immunology 91, 421–429 (1997).
Gregorova, M. et al. Post-acute COVID-19 associated with evidence of bystander T-cell activation and a recurring antibiotic-resistant bacterial pneumonia. Elife 9, e63430 (2020).
Habel, J. R. et al. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A*02:01 phenotype. Proc. Natl Acad. Sci. USA 117, 24384–24391 (2020).
Phetsouphanh, C. et al. Maintenance of functional CD57+ cytolytic CD4+ T Cells in HIV+ Elite Controllers. Front. Immunol. 10, 1844 (2019).
Lew, R. A. et al. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp. Physiol. 93, 685–693 (2008).
Acknowledgements
We thank the staff at the St Vincent’s Institute for Applied Medical Research–Clinical Trials Unit for their expertise in specimen processing and biobanking. We thank E. Johansson Beves for assistance with Cytek Aurora and V. Klemm for proofreading the manuscript. We appreciate grant support from the St Vincent’s Clinic Foundation, the Curran Foundation, the Rapid Response Research Fund and the Medical Research Futures Fund (Australia). S.K.P. and L.M.B. were supported by National Health and Medical Research Council (NHMRC) program grant APP 1055214 (L.M.B.), Medical Research Future Fund award GNT 1175865 and an Austin Medical Research Foundation grant. D.B.W. was supported by NSF-DMA grant 1902854. S.J.K. and J.A.J. were supported by the Victorian Government, MRFF award 2005544, NHMRC program grant 1149990 (S.J.K. and A.D.K.) and NHMRC fellowships (1136322 to S.J.K. and 1123673 to J.A.J.).
Author information
Authors and Affiliations
Contributions
C.P. and D.D. contributed to the experimental design, collected patient data, performed data experimentation and interpretation and drafted the manuscript. D.B.W. performed the log-linear classification model and statistical analysis. A.H. and C.P. performed and conducted experiments. C.M.L.M. provided unexposed serum and drafted the manuscript. S.K.P., J.A.J. and L.M.B. performed ACE2 experiments and interpreted data. G.J.D., S.J.K., A.D.K. and G.V.M. contributed to the experimental design and supervision of the study. C.P., A.D.K. and G.M. finalized the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Nature Immunology thanks Stuart Weisberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Ioana Visan was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 No difference in levels of 22 serum analytes following SARS-CoV-2 infection at month 4.
a, No differences in IL-1b, IL-6, TNF-α, IL-12p70, IFN-α2, IFN-λ2/3, GM−CSF, IL-10, IFN-γ, TGF-β1 or IL-13 levels between the 4 groups (LC, MC, HCoV and UHC) at 4 months. b, No differences in sCD25, PTX3, CCL2(MCP-1), IL-33, PD-1, IL-5, IL-9, MPO, PECAM-1, ICAM-1 or VCAM-1 levels between the 4 groups at 4-months. Data shown as medians with interquartile ranges. c, Median and IQR values for the 4 groups at 4 months. LC (long COVID), MC (asymptomatic matched controls), HCoV (human endemic coronavirus infected), UHC (unexposed healthy controls).
Extended Data Fig. 2 Reduction of some cytokines at month 8.
a, IFN-β and IFN-λ1 levels remained higher in LC at 8-months compared to HCoV and UHC; MC was only higher than HCoV at 8-months. b,c, Reduction of most analytes at 8-months post-infection. Data shown as medians with interquartile ranges Kruskal-Wallis with Dunn’s correction was used for 4-month analysis, p values <0.05 (*), <0.01 (**), <0.001 (***), <0.0001 (****), were considered significant. Wilcoxon t-test was used for paired comparison of 4- and 8-month data and Mann-Whitney t-test was used for unpaired samples. LC (Long COVID), MC (asymptomatic matched controls), HCoV (human endemic coronavirus infected), UHC (unexposed healthy controls); 4 M (4-months), 8 M (8-months).
Extended Data Fig. 3 Clustering of cell phenotypes.
a, Example of cell cluster phenotype identification using CellExplorer plugin on FlowJo. Cluster 14 is equivalent to population 3, identified as CD127lowTIM-3negCCR7+CD45RA+CD27+naïve CD4+ T cells. b,c, TriMap of T cell, B cell, NK Cell and myeloid cell clusters (outlined in red) at 3 months (b) and 8 months (c).
Extended Data Fig. 4 Deciphering naive phenotypes.
a,c,Representative TriMap and phenograph clustering of CCR7+CD45RA+ naïve T cells (a) and IgD+CD27− naïve B cells (c). Red gates and red text represent un-activated naïve subsets absent in LC. b,d, Phenotype and cell frequencies contributing to each cluster from naïve T cells (b) and naïve B cells (d). e,f, Representative histogram of CD127 expression on differing naïve T cell (e) and naïve B cell (f) populations (all data combined). Red text highlights subsets absent in LC. LC (long COVID), and MC (asymptomatic matched controls), UHC (unexposed healthy controls).
Extended Data Fig. 5 Activation markers expressed on mDCs and T cells.
a, No difference in activated state of Myeloid dendritic cells (mDCs) at 3 months and 8 months. b, No difference between LC and MC at 3 and 8 months when co-expression of PD-1 and Tim-3 were examined.Data shown as medians with interquartile ranges. Two-tailed p values <0.05 (*) were considered significant, ns (not significant). Wilcoxon t-test was used for paired samples and Mann-Whitney t-test was used for unpaired samples. LC (long COVID), and MC (asymptomatic matched controls), UHC (unexposed healthy controls).
Extended Data Fig. 6 Gating strategy.
a, Representative pseudo colour dot plot and gating strategy for flow cytometry analysis. Cells use for downstream analysis were live singlets expressing lineage markers; mDC (myeloid dendritic cells), pDC (plasmacytoid dendritic cells).
Supplementary information
Rights and permissions
About this article

Cite this article
Phetsouphanh, C., Darley, D.R., Wilson, D.B. et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol 23, 210–216 (2022). https://doi.org/10.1038/s41590-021-01113-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-021-01113-x
This article is cited by
-
Bulk RNA sequencing for analysis of post COVID-19 condition in adolescents and young adults
Journal of Translational Medicine (2024)
-
Characterisation, symptom pattern and symptom clusters from a retrospective cohort of Long COVID patients in primary care in Catalonia
BMC Infectious Diseases (2024)
-
Cardiovascular effects of the post-COVID-19 condition
Nature Cardiovascular Research (2024)
-
Post-COVID-19 conditions in children and adolescents diagnosed with COVID-19
Pediatric Research (2024)
-
Incident allergic diseases in post-COVID-19 condition: multinational cohort studies from South Korea, Japan and the UK
Nature Communications (2024)
