
Abstract
Development of effective human immunodeficiency virus 1 (HIV-1) vaccines requires synergy between innate and adaptive immune cells. Here we show that induction of the transcription factor CREB1 and its target genes by the recombinant canarypox vector ALVAC + Alum augments immunogenicity in non-human primates (NHPs) and predicts reduced HIV-1 acquisition in the RV144 trial. These target genes include those encoding cytokines/chemokines associated with heightened protection from simian immunodeficiency virus challenge in NHPs. Expression of CREB1 target genes probably results from direct cGAMP (STING agonist)-modulated p-CREB1 activity that drives the recruitment of CD4+ T cells and B cells to the site of antigen presentation. Importantly, unlike NHPs immunized with ALVAC + Alum, those immunized with ALVAC + MF59, the regimen in the HVTN702 trial that showed no protection from HIV infection, exhibited significantly reduced CREB1 target gene expression. Our integrated systems biology approach has validated CREB1 as a critical driver of vaccine efficacy and highlights that adjuvants that trigger CREB1 signaling may be critical for efficacious HIV-1 vaccines.
Similar content being viewed by others


Main
Despite decades of intensive research, HIV-1 remains at the forefront of human pathogens for which no effective vaccine has been developed. The RV144 trial, which is so far the most successful trial of an HIV-1 vaccine, used priming with the recombinant canarypox vector ALVAC (vCP1521) followed by boosts with ALVAC + AIDSVAX B/Egp120 that resulted in an estimated vaccine efficacy of 31.2% against HIV-1 infection1. The ALVAC prime was critical for the observed efficacy in RV144 as AIDSVAX administration alone did not induce protective humoral immunity in other vaccine trials2,3.
Studies showed that titers of non-neutralizing antibodies including V1V2-specific binding antibodies and IgG antibodies against linear epitopes in V2 and V3 of gp120 were correlates of decreased HIV-1 risk in the RV144 trial4,5,6,7,8. Studies in NHPs confirmed that the vaccine regimen used in RV144 induced protection: from low-dose mucosal challenge with the simian immunodeficiency virus SIVmac251; when Alum, but not MF59, was used as the adjuvant; and when ALVAC, not Ad26, was used as the prime9,10,11. Recent findings from HVTN702, the South African follow-up HIV vaccine trial to RV144 that used ALVAC + MF59, showed no reduction in acquisition of HIV-1 in vaccinated participants12. Understanding the cellular and molecular mechanisms of ALVAC- and Alum-mediated vaccine efficacy is critical for understanding the poor protective responses observed in the HVTN702 trial.
Monocytes/macrophages and dendritic cells (DCs) are known targets of ALVAC and are critical modulators of innate immune responses13,14. Activated innate immune cells produce cytokines/chemokines that drive the migration and differentiation of leukocytes, including CD4+ T cells and B cells, from the blood to the site of vaccination15. We performed an NHP immunogenicity study (Study 36) to delineate the function of ALVAC priming in mediating efficacious vaccine-elicited immune responses. Our studies highlight the role of the CREB1 pathway and an associated cytokine/chemokine network in mediating the protection observed in the RV144 trial.
Results
ALVAC-induced CREB1 activity predicts reduced HIV-1 infection
To define the immune mechanisms triggered by ALVAC priming in Study 36 (schematic in Extended Data Fig. 1a), transcriptional profiling of purified DCs, CD4+ T cells and B cells was performed at pre-vaccination, and days 2/3, week 2 and week 25 post initial vaccination in NHPs immunized with ALVAC-HIV-1 (G2) or empty ALVAC (G1). The sample collection and analytical strategy used herein is detailed in Extended Data Fig. 1a,b. Gene set enrichment analysis (GSEA)16 was used to analyze gene expression datasets at all time points post-vaccination. Induction of type I and type II interferons (IFNs) and IFN-stimulated genes has been shown to promote protective immune responses in vaccines17,18,19. While IFN signaling was induced in DCs and CD4+ T cells at days 2/3 post-vaccination (when immunization was performed in both G1 and G2 arms), IFN pathway induction did not persist and more importantly it was not significantly correlated to V1V2 IgG titers (referred to as V1V2 throughout this manuscript) at all time points collected in our study (r and P values in Extended Data Fig. 2).
Using GSEA, we identified chromatin immunoprecipitation sequencing (ChIP–seq)-confirmed transcription factor (TF) target gene sets (ChEA database20) that: were induced by ALVAC in all three cell subsets at each of the three time points independently in the G1 and G2 arms compared to pre-vaccination; and consistently correlated in all three microarray time points to all four time points where V1V2 titers were assessed (54 total contrasts in Supplementary Table 1). We identified 62 TF target gene sets that were significantly induced by ALVAC in all three cell subsets across all three time points and 45 TF gene sets that significantly correlated with V1V2 across all four time points (Fig. 1a, statistics in Supplementary Table 2). Forty-two of these TF gene sets were identified as ALVAC induced and as correlates of V1V2 magnitude and durability. These TFs represent putative drivers of ALVAC-mediated vaccine responses.

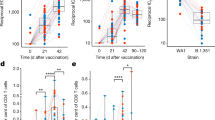
a, Venn diagrams of TF target gene sets that are ALVAC induced at all 3 post-vaccination time points in DCs, CD4+ T cell and B cells (n = 5 G1 and 5 G2 NHPs per subset) and correlate positively to V1V2 titers measured at four time points. A total of 62 TF gene sets were persistently ALVAC induced and 45 TF genes sets consistently correlated to V1V2 titers; 42 in common to both. To confirm these gene sets were also correlated to V1V2 titers and reduced HIV-1 acquisition in RV144, we used microarray data from the RV144 case–control study5,22 that down-sampled the RV144 cohort at a ratio of 5:1 uninfected versus infected. b,c, The 42 TF target gene sets identified in a were then tested for ALVAC induction (n = 24 participants in RV144 pilot trial) of gene expression at 2 weeks post-final vaccination compared to pre-vaccination (b) and correlation of gene expression with V1V2 titers, both at 2 weeks post-final vaccination (n = 182 RV144 participants; c). CREB1 was the second most potently induced TF gene set and the fourth highest correlate of V1V2 titers, highlighting a novel role for CREB1 in RV144. NES, normalized enrichment score. d, In RV144, vaccinated individuals who do not become infected with HIV (n = 151 RV144 participants) have significantly elevated CREB1 target gene expression (z-score) compared to those who do become infected (n = 31) using logistic regression, corrected for sex and baseline behavioral risk (****P = 0.00095). The limits of the box represent Q1 (lower) and Q3 (upper), and the middle line shows the median. Error bars represent 1.5 × IQR. e, Using a log-rank test, CREB1 z-scores were split into tertiles (low = 61, medium = 61, high = 60) revealing that the medium and high CREB1 tertiles have significantly reduced HIV acquisition compared to the low tertile, with the highest tertile showing sustained lowered risk up to 36 months post vaccination.
To associate these TF gene sets with reduced risk of HIV acquisition within 42 months of vaccination, we investigated microarray data from the RV144 case–control study4. We probed the RV144 data to determine whether the 42 TF gene sets (identified above) were induced post-vaccination with ALVAC and correlated with V1V2 (Fig. 1b,c). We observed that the CREB1 target gene set was unique as it was the second most potently induced gene set (post-ALVAC) and fourth ranked correlate of V1V2 (Fig. 1b,c). Importantly, CREB1 target genes included a substantial number encoding cytokines/chemokines (and their receptors) known to drive immune modulatory functions (see Supplementary Table 3 for full list of CREB1 target genes). Among the top ten gene sets for each contrast, we found four other TF target gene sets that were also induced by ALVAC and correlated with V1V2. However, these four TFs are not highly related to innate or adaptive immune functions but rather regulate general cellular processes of: cell growth/differentiation (THAP11)21, heart development (TBX5)22, pancreas function (PDX1)23 and cell cycling (CCND1)24. Furthermore, these four TFs were not consistently identified as the top TFs across all subsets in Study 36 (values in Supplementary Tables 1–6).
The average expression of leading-edge genes (LEGs) from the CREB1 target gene set (CREB1 z-score) was significantly elevated in vaccinated participants who were not infected by HIV-1 compared to participants who were infected post-vaccination (Fig. 1d). Kaplan–Meier analysis of RV144 trial participants separated into three groups (tertiles) based on CREB1 z-score showed significantly reduced risk of HIV-1 acquisition in the medium and high CREB1 z-score tertiles (Fig. 1e). Furthermore, participants with high CREB1 z-scores maintained a substantially lower risk of acquisition (P = 0.0548) up to 3 years after vaccination compared to the medium tertile (Fig. 1e). To validate our identification of CREB1 by GSEA in Study 36, we performed hypergeometric means testing as an independent approach to identify contrasts with significant enrichment for CREB1 target genes. In line with our GSEA Study 36 findings, 35 of 36 contrasts for ALVAC induction and V1V2 correlation, comprising each time point and each subset in Study 36, showed significant enrichment of CREB1 target genes (Extended Data Fig. 2c). Our data highlight that ALVAC-induced CREB1-driven genes significantly discriminate participants that do not acquire HIV-1 post-vaccination in the RV144 trial and have protective V1V2 antibody responses in both Study 36 and RV144 trials.
To monitor the kinetics of induction of CREB1 target genes post ALVAC vaccination, we analyzed the transcriptome of peripheral blood cells at 16 h, 24 h, 48 h and 72 h after vaccination for the 4 immunizations (administered at weeks 0, 4, 12 and 24 of the study) in 6 NHPs from an independent study. The CREB1 target gene signature was downregulated at 16 h possibly due to the extravasation of cells from the blood into tissue and lymphoid organs immediately following vaccination. However, CREB1 target gene expression was significantly enhanced at 24 h and continued to increase up to 72 h after vaccination (Extended Data Fig. 3a). The Jaccard index was used to compare the overlap of LEGs for each of these time points; we observed significant overlap between the LEGs of time points showing positive CREB1 target gene induction (thick lines between red nodes) but limited overlap was noted between negative time points (thin lines between blue nodes; Extended Data Fig. 3b). These results indicate that the set of genes negatively modulated by CREB1 at 16 h are distinct from those positively modulated at 24–72 h. Our findings demonstrate that CREB1-driven genes are induced early post-immunization with ALVAC, and that this induction persists over time (up to 3 days) across consecutive vaccinations, highlighting the central role for CREB1 in modulating HIV-1 vaccine responses.
CREB1-associated cytokines augment migration and V1V2 titers
Pox vectors are potent inducers of cytokines and chemokines25. We investigated the impact of ALVAC-induced CREB1 target genes on cytokine/chemokine expression; transcriptional profiling identified 103 cytokines, chemokines and growth factors that were ALVAC induced and correlated with V1V2 in at least one cell subset and time point in Study 36 (Supplementary Table 5). To confirm that induction of these cytokines post-ALVAC immunization was associated with higher V1V2, we quantified the plasma levels of 23 of these cytokines/chemokines (using a custom cytokine array) at weeks 4 and 27 post-immunization in Study 36. Univariate analysis revealed that the chemokines fractalkine (CX3CL1), GROα (CXCL1) and MCP1 (CCL2) and the cytokines FLT3LG and transforming growth factor-β1/3 (TGF-β1/3) were significant positive correlates of V1V2 (Extended Data Fig. 4a). In contrast, eotaxin 3 (CCL26), interleukin-13 (IL-13), IP10 (CXCL10) and IL-6 were negative correlates of V1V2 titers (Extended Data Fig. 4a).
Networks of multiple cytokines are required to trigger effector arms of immunity during vaccination. We used Lasso regression26 to model a signature of cytokines that shows better predictive value for V1V2 than each individual cytokine or chemokine (Extended Data Fig. 4b). The week-4 plasma signature predictive of V1V2 at week 53 included the chemokines fractalkine (CX3CL1: positively associated), eotaxin 3 (CCL26: negatively associated) and I-TAC (CXCL11: negatively associated) and the cytokines TGF-β3 (positively associated) and IL-13 (negatively associated). The week-27 plasma signature predictive of V1V2 at week 55 consisted of the chemokines GROα (CXCL1) and MCP1 (CCL2) and the cytokine IL-18, all positive correlates (Extended Data Fig. 4b). Using GeneMania, we built a network that inferred the top 30 neighboring genes associated with the cytokines identified in our multivariate model (Fig. 2a). Of these 30 genes, 27 were cytokine/chemokine-encoding genes that are known targets of CREB1 and/or have been transcriptionally identified as correlates of V1V2 in Study 36. When z-scores for the network in Fig. 2a were split into tertiles in RV144, these cytokines and chemokines were positively associated with reduced HIV-1 acquisition, although not as strongly as the entire CREB1 target gene set (Extended Data Fig. 4c). Our results indicate that these CREB1-associated cytokines/chemokines are critical immune modulators that drive efficacious HIV-1 vaccine responses. In line with the known role of G-protein-coupled receptor (GPCR) signaling in promoting chemokine-driven cell migration, we observed that DCs, CD4+ T and B cells showed enhanced expression of chemotaxis and GPCR signaling pathways post-vaccination in Study 36 (Extended Data Fig. 4e).

a, A GeneMania network of cytokine correlates from the Study 36 Lasso model with the top 30 closest associated proteins (criteria in figure legend). This network reveals that these key plasma cytokines are associated with a larger network of cytokines/chemokines that are induced during ALVAC vaccination and correlate to V1V2 titers. Circles represent cytokines detected at week 4 that correlate to week-53 titers, and squares represent cytokines at week 27 that correlate to week-55 titers. Black filled nodes are the products of genes identified as correlates of V1V2 in our transcriptional analysis. b, To confirm that our findings directly linked with protection from SIV/HIV challenge, we probed data from the P162 vaccine study, an ALVAC-SIV + Alum immunization study (n = 26 independent NHPs) that used the RV144 regimen followed by up to ten intrarectal SIV challenges post-vaccination. NHPs were split into tertiles (low = 9, medium = 9, high = 8) based on the CREB1 pathway z-score using a log-rank test. This analysis reveals a substantial increase in protection (number of challenges until infected) between the CREB1 z-score high and low tertiles. Mean challenges to infection: low = 2.56, medium = 3.72, high = 4.82. c, The CREB1 target gene set from ChEA shows significant positive enrichment for genes that correlate to rectal IgG including that encoding cV2 using preranked GSEA (adjusted P < 0.002 for all). d, Monkeys that produced cV2 in P162 (n = 18) have significantly elevated (P = 0.0087) CREB1 target gene expression (z-score) compared to cV2− (n = 8) using a two-sided Wilcoxon test (**P = 0.001). The lower limit of the box represents Q1, the upper limit represents Q3, and the middle line shows the median. The top and bottom of the error bars represent 1.5 × IQR. e, Levels of circulating fractalkine (CX3CL1) are a significant positive correlate, using Spearman correlation, of number of challenges until infection within cV2+ NHPs while eotaxin 3 (CCL26) is a negative correlate of challenges in cV2+ monkeys (n = 12 for which measures were taken).
We next assessed the association between key cytokines/chemokines, and their receptors, with V1V2 among the three immune cell subsets in Study 36 (Extended Data Fig. 4f). Of the 18 cytokines/chemokines identified transcriptionally (along with their specific receptors), 4 were expressed by DCs and CD4+ T cells and 4 were expressed by all 3 subsets. Analysis of receptor expression revealed similar findings with the majority (9/13) expressed commonly in DCs and CD4 + T cells (6), or in all 3 subsets (3) (Extended Data Fig. 4f). Key pairs of cytokines/chemokines and their receptors (identified in Extended Data Fig. 4a,b) included CX3CL1/CX3CR1, IL-18/IL-18R1, TGF-β1/TGF-βR and FLT3LG/CD135 (also known as FLT3R). We further showed that 18 chemokines in DCs or CD4+ T cells at weeks 0/2 (including fractalkine (CX3CL1)) correlated with chemotaxis pathways in at least 1 of DCs, CD4+ T cells or B cells (Supplementary Table 6). These data demonstrate that plasma levels of CREB1-associated cytokines/chemokines predict the magnitude of V1V2 and are key members of a larger network of cytokines and chemokines that potentiate induction of protective immunity post-vaccination by triggering migration and chemotaxis that is associated with protection from acquisition.
The CREB1 signature drives protection from challenge in NHPs
We next monitored for the association of CREB1 induction by ALVAC in conferring protection from challenge in the P162 study10. This independent ALVAC-SIV NHP study, which uses the same vaccination regimen as the RV144 trial, assessed vaccine efficacy by administering rectal SIV challenges to the NHPs. Ranking of vaccinated and challenged NHPs into tertiles based on their CREB1 z-score generated from transcriptional profiling of their PBMCs at post second boost (week 25) showed that NHPs with the highest CREB1 z-score exhibited enhanced protection from challenge (P = 0.057) compared to NHPs in the lowest tertile; importantly, all four NHPs that were protected from ten challenges were in the high CREB1 tertile (Fig. 2b). We next correlated the expression of CREB1 target genes post-vaccination in P162 with rectal anti-SIV IgG antibodies, namely cV2, which was previously shown to be a surrogate marker of protection from challenge in this study10. The CREB1 target gene set showed significant positive enrichment for genes that correlated to rectal IgG against cV2, as well as other rectal SIV-specific IgGs (Fig. 2c). We also observed significantly higher CREB1 z-scores (P = 0.001) in cV2-positive NHPs compared to cV2-negative NHPs (Fig. 2d).
In addition, we observed a significant positive correlation between the levels of fractalkine (CX3CL1), an inducer of monocyte migration27, in plasma at week 5 and the number of challenges needed to infect NHPs producing cV2-specific antibodies (Fig. 2e). We found that the levels of eotaxin 3 (CCL26), a repressor of monocyte migration28, were a negative correlate of the number of challenges in cV2-positive NHPs (Fig. 2e). This confirms our data in Study 36 where fractalkine was positively associated with protective V1V2 while eotaxin 3 was negatively associated with V1V2. Together, data from P162 confirm our findings in Study 36 and RV144 identifying the critical role of CREB1; these results directly show that the CREB1 pathway is linked with protection from HIV/SIV acquisition following ALVAC vaccination.
CREB1 orchestrates subset-specific transcriptional networks
We next probed for the specific immune functions (in sorted DC, CD4+ T cell and B cell subsets) associated with CREB1 transcriptional activity in Study 36. We identified TF-regulated gene sets in the ChEA database that correlated to the CREB1 z-score and positively correlated to V1V2 (P < 0.05), generating a list of CREB1-associated TF target gene sets in DCs, CD4+ T cells and B cells, independently. Using ClueGO, we identified genes that encode specific biological processes and tested their enrichment in curated pathways from GO and REACTOME using two-sided hypergeometric testing (P < 0.05). Overlapping pathways were then collapsed based on significant overlap in LEGs (P < 0.05) to generate functional nodes and their associated genes. The ClueGO network for DCs was comprised of CREB1 LEGs and CREB1-associated TF LEGs that regulate nodes of genes associated with lymphocyte differentiation/activation and migration including those encoding the known regulators of DC function FLT3LG (drives DC development29), IL-18 (T cell differentiation30), BCL3 (promotes T cell priming31 and differentiation of type 2 helper T (TH2) cells32) and TNFSF4 (also known as OX40L; T cell costimulation33). Of note, FLT3LG and IL-18 were previously identified as plasma correlates of V1V2 in Study 36. TMEM173 (STING) and TBK1, known sensors of pathogen34,35 and host36 DNA leading to induction of antiviral signaling, were central CREB1-driven genes associated with the nucleic acid–sensing nodes (Fig. 3a).

To understand how CREB1 drives a master transcriptional network associated with efficacious vaccine responses, TF target gene sets that correlated positively with V1V2 titers and CREB1 target gene set z-scores were identified. Genes from these CREB1-associated TF gene sets were combined with the LEGs of the CREB1 target gene set. Genes in these networks were then annotated to functional pathways and nodes using ClueGO, an application in Cytoscape. a, CREB1 and its associated gene sets in DCs drive functional nodes of lymphocyte/leukocyte migration, leukocyte differentiation and cytosolic nucleic acid sensing (STING/TMEM173). b, ClueGO analysis of CREB1 and associated gene sets in CD4+ T cells highlights multiple critical nodes for CD4+ function including costimulation, cytokine signaling, migration and antigen processing/presentation. c, A ClueGO network highlighting enrichment of CREB1 and its associated gene sets in pathways of migration in B cells. d, CREB1-associated TF gene sets were identified in RV144 followed by identification of subsets from Study 36 in which CREB1 associated with each TF. This analysis confirms that CREB1 associates with a range of TFs involved in immune function in both RV144 and Study 36, and that these associations are cell-type specific.
In CD4+ T cells, the CREB1-associated ClueGO network included nodes with genes involved in cytokine signaling, costimulation/activation and antigen processing/presentation (Fig. 3b). We found key genes with known T cell function as central drivers of these nodes including those encoding CD4, CD247 (CD3ξ) and AKT1 as well as the cytokines/receptors FLT3LG, IL-24 (associated with TH2 responses37) and TNFRSF14 (T cell growth/survival38). Analysis of the B cell ClueGO network revealed nodes of leukocyte and lymphocyte migration genes including those encoding CCR7, DOCK8 (involved in chemokine GPCR signaling39) and SPN (CD43))40 (Fig. 3c). To demonstrate that CREB1 modulates a similar master transcriptional network in RV144 as was seen in Study 36, we identified TF gene sets in RV144 that correlated with CREB1 z-score and V1V2, the most significant immunological surrogate of reduced HIV-1 acquisition (Fig. 3d). Importantly, all of these TFs (with the exception of NUCKS1) were identified as associated with CREB1 z-score in at least one cell subset from Study 36 and are themselves known or predicted targets of CREB1. These data confirm the novel association between CREB1 and key TFs that drive transcriptional networks associated with reduced HIV-1 acquisition, potentiate migration of all three immune subsets and promote subset-specific effector functions.
ALVAC induces CREB1 binding in the promoter of target genes
To provide direct mechanistic evidence of ALVAC-induced CREB1 binding to key target genes, we performed CREB1 ChIP–seq on PBMCs from naive NHPs infected in vitro with empty ALVAC for 24 and 48 h or ALVAC-HIV for 24 and 48 h. We identified 4,778 genes that exhibited enriched CREB1 binding within 3 kilobases (kb) of their transcription start site (TSS) in at least 1 of the 4 conditions compared to medium alone. A total of 1,307 of these genes overlapped with the ChEA CREB1 dataset (Extended Data Fig. 5a) and significant overlap of genes was observed between all 4 infection conditions using the SuperExactTest41 (Extended Data Fig. 5b). These genes included those encoding the chemokines fractalkine (CX3CL1) and CCL2 (MCP1), previously identified by gene expression profiling and plasma quantification in Study 36, and the antigen presentation gene encoding Mamu-E (homolog of HLA-E; Extended Data Fig. 5c). To validate our findings in Study 36, we used SuperExactTest and observed significant overlaps between the LEGs in Study 36 and ChIP–seq genes for DCs, CD4+ T cells and B cells (up to 646, 560 and 554 genes, respectively) confirming that ALVAC directly induces CREB1 binding to genes identified as transcriptional correlates of V1V2 (Fig. 4a,b and Extended Data Fig. 5d). We used GeneMania to visualize the network of immune-related genes that were in common to our ChIP–seq results and one or more subsets in Study 36. Among these overlapping genes are those encoding CXCL11, FOS, JUN, HLA-E and HLA-DOA (Fig. 4c).

a–e, PBMCs from two healthy NHP donors (a–c) or two healthy human donors (d,e) were infected in vitro with a multiplicity of infection of 10:1 of ALVAC-HIV or empty ALVAC, or left in medium. Cells were collected after 24 and 48 h and fixed, and ChIP–seq was performed for CREB1. Genes were identified that showed significantly enriched CREB1 binding within 3 kb of their TSS in ALVAC infection compared to medium. a,b, Bar plots of the overlap between genes showing enriched CREB1 binding by ChIP–seq for one or more of the ALVAC infection conditions and the LEGs that were ALVAC induced and correlated with V1V2 titers in Study 36 in DCs (a) and CD4+ T cells (b). Significant overlap is observed for all combinations tested. c, A GeneMania network of immune-related CREB1 target genes that overlap between ALVAC-induced genes by ChIP–seq and at least one cell subset in Study 36, annotated for the subset(s) they overlap with. d, A bar plot showing significant overlap between human ChIP–seq genes, the LEGs from RV144 and the LEGs from DCs, CD4+ T cells and B cells in Study 36. Significant overlap is seen for all combinations, including 45 in common to all, demonstrating there are CREB1 target genes that are conserved across all studies. e, A table showing nominal P values and NESs of GSEA using custom gene sets made from ChIP–seq and transcriptional overlaps. This analysis shows that ChIP–seq-confirmed genes predict reduced HIV-1 acquisition in RV144 equivalent to the entire CREB1 ChEA gene set. SuperExactTest was used for this as it was designed specifically to identify statistically significant overlap in multi-set interaction analysis among three or more sets.
To directly associate ALVAC-induced CREB1 activity with reduced HIV-1 acquisition, we performed CREB1 ChIP–seq on human PBMCs infected with empty ALVAC or ALVAC-HIV for 24 h. We identified genes with key immunological functions that were enriched for CREB1 binding after ALVAC infection including those encoding FLT3LG, HLA-DMB and TBK1 (Extended Data Fig. 5e). A total of 1,902 genes overlapped with the ChEA CREB1 target gene set (Extended Data Fig. 5f). Significant overlap was observed between our human ChIP–seq data and the CREB1 LEGs from the RV144 trial, including 352 genes in common with reduced HIV-1 acquisition and 108 genes in common to all 3 contrasts from RV144 (Extended Data Fig. 5g). Among these 108 genes are those encoding CREB1 itself and key immunoregulatory genes encoding FOS, REL, RELB, IRF1, MAPKs, STAT5B and BCL6. We observed significant overlap between our human ChIP–seq results and all of the LEGs from RV144 and Study 36, with 45 genes in common to all comparisons in RV144 and Study 36 including those encoding MAPK signaling regulators (NELL2 (also known as NRP2), MAPK3K10 and MAPKAPK5; Fig. 4d). In fact, these 45 genes predicted reduced HIV-1 acquisition in RV144 as accurately as the entire ChEA CREB1 dataset (P = 0.00022 versus P = 0.00063, respectively), clearly showing that there are conserved CREB1 target genes across NHPs and humans that are ALVAC induced and associated with reduced HIV-1 acquisition (Fig. 4e). These findings validate CREB1 as a critical TF induced by ALVAC vaccination that drives expression of key immune genes that modulate reduced HIV-1 acquisition post-vaccination.
The adjuvant MF59 reduces the CREB1 signature compared to Alum
The HIV-1 vaccine trial HVTN702 was stopped due to lack of efficacy in preventing HIV-1 acquisition12. The P162 NHP trial used for analysis in this study compared the efficacy of the ALVAC regimen used in RV144 (ALVAC + Alum) to the one used in HVTN 702 (ALVAC + MF59)10. To address whether the CREB1 target gene signature was differentially induced by these two adjuvants, we generated CREB1 z-scores using the LEGs from the MF59 versus Alum contrast on the PBMC microarray data following the third immunization. We observed significantly increased CREB1 z-scores in NHPs immunized with ALVAC + Alum compared to NHPs immunized with ALVAC + MF59 (P = 1.2 × 10−6; Fig. 5a). We next used GSEA to interrogate whether chemotaxis pathways and GPCR signaling were altered in MF59 compared to Alum-adjuvanted NHPs. Figure 5b shows that pathways of chemotaxis and GPCR signaling were significantly enriched in the Alum arm compared to MF59, including five pathways involved in leukocyte migration and GPCR signaling (asterisks in Fig. 5b) previously identified as ALVAC induced and correlates of V1V2 in Study 36 (Fig. 5b). Moreover, several immune-related pathways were significantly enriched (false discovery rate < 0.05) in Alum compared to MF59 in P162, which also correlated with reduced HIV-1 acquisition in RV144 (Fig. 5c and Extended Data Fig. 6a). The intersection of genes found to be both increased in Alum and negatively associated with acquisition in RV144 was used to generate a per participant z-score for each common pathway. These scores were then split into tertiles for log-rank comparison of HIV-1 acquisition (Fig. 5d,e). These pathways include DC maturation and antigen processing (Fig. 5d) as well as signal transduction pathways upregulated in monocytes/macrophages post stimulation with LPS/IFNs (Fig. 5e); these pathways showed enrichment of CREB1 target genes that collectively correlated with reduced HIV-1 acquisition (genes in Extended Data Fig. 6b). Of note, most of these genes map to pathways of innate immune responses. These results link functional immune pathways augmented in Alum versus MF59 adjuvantation with reduced HIV-1 acquisition in RV144, providing a potential mechanism underlying the lack of efficacy observed in HVTN702.

a, In P162, NHPs treated with ALVAC + MF59 (n = 27 independent NHPs) show significantly reduced (****P = 1.2 × 10−6) CREB1 target gene activation compared to ALVAC + Alum-treated NHPs using logistic regression. The lower limit of the box represents Q1, the upper limit represents Q3, and the middle line shows the median. The top and bottom of the error bars represent 1.5 × IQR. b, Pathways of chemotaxis were significantly upregulated in Alum-treated NHPs, including pathways identified in Study 36 (annotated by an asterisk). c, Immune-related pathways that are significantly enriched in Alum versus MF59 and negatively correlate to HIV acquisition in RV144 are identified using GSEA. d,e, Splitting of pathway z-scores into tertiles using a log-rank test confirms the significant reduction in HIV acquisition for pathways of DC maturation and antigen processing/presentation (d) and pathways of innate immune activation and IFN signaling in monocytes/macrophages (e).
cGAMP induces p-CREB1 activation and cytokine production
Results of Study 36 (Fig. 3a) linked nucleic acid sensing and the genes encoding TMEM173 (STING) and its kinase TBK1 with CREB1 in DCs. In line with a role for HIV DNA in inducing cGAS–STING-mediated responses42,43, we observed a significantly higher number of CREB1-induced genes in the G2 (ALVAC-HIV) arm compared to G1 (empty ALVAC) suggesting that sensing of both ALVAC and HIV DNA contributes to the activation of CREB1 signaling (Supplementary Fig. 6c). We hypothesized that ALVAC-HIV could activate cGAS–STING in DCs leading to activation of CREB1 and cGAMP release from DCs and subsequent induction of paracrine CREB1 signaling in neighboring and infiltrating cells, including CD4+ T cells and B cells, explaining the ‘infectious’ CREB1 activation observed in Study 36 (ref. 44).
To test this hypothesis, we stimulated PBMCs from healthy donors with 1 and 10 μM cGAMP and quantified the presence of p-CREB1 Ser133 in innate and adaptive immune subsets using PhosFlow staining. We used unbiased RPhenograph analysis to identify clusters of cells (clusters visualized on a uniform manifold approximation and projection (UMAP) plot of all cells) differentially induced by 1 μM cGAMP compared to medium after 15 min of stimulation (Fig. 6a). We identified four clusters significantly induced by cGAMP: cluster 5 (P = 0.0095), cluster 16 (P = 0.0126), cluster 17 (P = 0.0288) and cluster 20 (P = 0.0063). A row-normalized heatmap, showing the median fluorescence intensity (MFI) for each marker per cluster, revealed that the four clusters induced by cGAMP are marked by heightened expression of p-CREB1 and fell in the CD4+ T cells and CD19+ B cell populations (Fig. 6b). These results were corroborated by density plots and p-CREB1 MFI overlays onto UMAP clusters that showed regions of increased cellular density in cGAMP-treated samples that expressed high levels of p-CREB1 (Fig. 6c,d). Importantly, this cGAMP-induced p-CREB1, which is evident at 15 and 30 min, is reduced to unstimulated levels by 60 min—indicative of the rapid on/off kinetics of canonical phospho-signaling (Extended Data Fig. 7c). Our results were confirmed by manual analysis, using FlowJo, where we observed significant increases in p-CREB1 MFI in immune subsets including CD4+ T cells, B cells and monocytes/DCs (Extended Data Fig. 7d,e). The same p-CREB1 induction and phospho-kinetics were observed by UMAP and manual analysis when stimulating PBMCs with 10 μM cGAMP (not shown and Extended Data Fig. 7d,e).

a–d, Healthy human PBMCs (n = 5) were stimulated with 1 μM and 10 μM cGAMP for 15, 30 and 60 min. a,b, UMAP and RPhenograph analysis identified clusters induced by treatment of PBMCs with 1 μM cGAMP for 15 min. Clusters 5, 16, 17 and 20 were identified as significantly induced by cGAMP treatment. b, A row-normalized heatmap of per-cluster marker (MFI) expression shows these significantly induced populations are p-CREB positive (*P < 0.05). c,d, Significantly modulated clusters were marked on density plots (c) and overlays (d) of p-CREB fluorescent intensity for each cell to visualize how areas of augmented cell density in cGAMP samples overlap with the areas of high p-CREB MFI (orange and red in d). These confirm that cGAMP treatment increases p-CREB. e,f, pan-DCs (n = 5) and CD4+ T cells (n = 4) were purified from healthy human PBMCs and stimulated with 10 μM cGAMP for 24 h followed by quantification of key cytokines correlating to V1V2 titers in Study 36. The two cell subsets were analyzed separately as well as combined as biologically distinct replicates. e, FLT3LG was significantly induced in combined purified DCs and CD4+ T cells (n = 9), with a substantial increase in CD4+ T cells alone (**P = 0.0097). Statistical analysis was performed using a two-sided, paired t-test. f, IL-18 was significantly induced in purified DCs with more potent induction observed when combined with CD4+ T cells (*P = 0.0360; **P = 0.0041). The center line indicates the median, the lower line represents the first quartile, and the upper line represents the third quartile. In the DC + CD4+ T cell graphs, the black symbols represent DCs and the blue symbols represent CD4+ T cells. The lower line in the violin plot represents Q1, the middle line shows the median, and the upper line represents Q3.
To link cGAMP-induced CREB1 with augmented immune function, we quantified production of key cytokines/chemokines, previously identified in Study 36, from purified pan-DCs and total CD4+ T cells following stimulation with 10 μM cGAMP. We found that FLT3LG was one of the most potently induced cytokines along with TRAIL (Extended Data Fig. 8a). FLT3LG was produced by purified CD4+ T cells (P = 0.0623) and by DCs (P = 0.1193) 24 h following stimulation with cGAMP, and this induction became highly significant (P = 0.0087) when data from the two cell subsets were combined, as distinct biological replicates, into one analysis (Fig. 6e). We also observed that cGAMP stimulation induced IL-18 in DCs (P = 0.0360) and CD4+ T cells (P = 0.1096) after 24 h, with strong significance (P = 0.0041) seen when the data from the two subsets were combined (Fig. 6f). Similar results were seen for three other CREB1-associated cytokines: GROα (CXCL1), TRAIL and TGF-β1 (Extended Data Fig. 8b). These data provide direct evidence that cGAMP can trigger the production of key cytokines and chemokines in DCs and CD4+ T cells that are driven by CREB1 activation and which were transcriptional and plasma correlates of V1V2 in Study 36. The combination of these in vitro experiments with our multi-omics data provides a novel mechanism by which ALVAC promotes immune function and vaccine efficacy (model in Extended Data Fig. 8c).
Discussion
In this study, we have identified a CREB1-induced signature that is a strong correlate of immunological outcomes and protection from SIV/HIV acquisition in NHPs and humans. We show that this new CREB1 signature is a more significant predictor of reduced HIV-1 acquisition than V1V2 and provides a more comprehensive understanding of the immunological mechanisms of protection. These results were obtained and validated in three independent cohorts (two NHPs and one human) using three different omics (RNA microarray/sequencing, ChIP–seq and multiplexed cytokine/chemokine detection). Our ChIP–seq results have validated the association between CREB1 target genes and reduced HIV-1 acquisition, identifying a core of 45 genes common to all studies that predict reduced HIV-1 acquisition equivalent to the entire CREB1 target gene set. While the specific CREB1 target genes associated with outcome showed variance across subsets and species, we observed a common set of CREB1-regulated TFs induced across these studies that drive genes that were validated by quantitative cytokine assessment in the plasma and CREB1-binding genes by ChIP–seq analysis in NHPs and humans. This highlights that ALVAC activates a common, global network of TFs that drives the cell-subset-specific immune responses required for efficacious vaccine responses and protection from HIV-1 acquisition.
Our findings led us to develop an integrated model of ALVAC- and CREB1-mediated HIV-1 vaccine efficacy. In step 1, DCs and monocytes at the site of immunization are infected with ALVAC-HIV. ALVAC and HIV DNA are sensed by cGAS42,43 leading to the production of the dicyclic nucleotide cGAMP. This is confirmed by our microarray data showing that STING signaling is associated with CREB1 activity in DCs in Study 36. CREB1 has previously been associated with cytokine production from DCs and in vitro activation of DCs45. Our studies show that this ALVAC-induced CREB1 is rapid (within 24 h) and persists temporally, highlighting the potential for this to function as a potent adjuvant to induce early and sustained vaccine responses.
In step 2, CREB1 activation drives the production of key cytokines and chemokines that generate chemotactic gradients that promote leukocyte migration to the site of vaccination46. Indeed, enhanced migration was the only common functional node associated with CREB1 activity in DCs, CD4+ T cells and B cells. We show that CREB1-driven chemokines produced by DCs and CD4+ T cells correlate positively with chemotaxis pathways in DCs, CD4+ T cells and B cells. In step 3, circulating leukocytes including DCs, CD4+ T cells and B cells extravasate from the blood into the site of vaccination and lymph nodes, where they migrate to sites of antigen encounter and presentation. The lack of conserved cytokine and chemokine upregulation across our studies could be due to differences in the individual biology of each subset of studies and in temporal kinetics when comparing samples from different time points post-vaccination. Indeed, our transcriptional and plasma cytokine analysis identified positive inducers of cell migration downstream of CREB1 (that is, fractalkine (CX3CL1))27 associated with vaccine immunogenicity and protection in two independent NHP studies, while repressors of monocyte migration (that is, eotaxin 3 (CCL26))28 were negative correlates of these outcomes. These data highlight that the balance of cytokines/chemokines induced and repressed post-vaccination is crucial for the recruitment of innate and adaptive immune cells and activation of the effector programs required for protective responses. This is further supported by our identification of TGF-β1/3 as positive correlates of V1V2 IgG highlighting a potential role for TGF-β in promoting Tfh responses required for optimal antibody responses.
In steps 4/5, leukocytes including CD4+ T cells recruited to the site of immunization/lymphoid tissue become activated by the release of cGAMP from ALVAC-infected cells. Our in vitro results reveal that cGAMP treatment induces p-CREB1 in immune cells and drives the production of cytokines/chemokines from purified DCs and CD4+ T cells. Two of these cytokines, FL3TLG and IL-18, were encoded by central CREB1-modulated genes driving functional nodes of T cell costimulation pathways, cytokine signaling and augmented antigen processing/presentation in CD4+ T cells. Our ChIP–seq results provide molecular validation of FLT3LG, IL-18 and MHC class I/II genes as CREB1 driven during ALVAC infection and show significant overlap in CREB1 target genes between NHPs and humans. Although the specific CREB1 target genes that were associated with V1V2 in Study 36 and reduced HIV-1 acquisition in RV144 showed partial but significant overlap, we found the upstream TFs that drove the transcription of these genes and were associated with CREB1 to be consistent across Study 36 and RV144. These results highlight that CREB1 drives a master transcriptional network that includes other TFs that is associated with efficacious HIV-1 vaccine outcomes in multiple immune subsets and across species. One potential mechanism explaining the infectious spread of CREB1 signaling is sensing of cGAMP by P2Y receptors that are known to be expressed on immune cells and sense extracellular nucleotides47. In Study 36, we found P2YR2 in DCs and P2YR1 and P2YR11 in CD4+ T cells as positive correlates of V1V2, with the last of these being a known driver of cAMP responses48.
The importance of the CREB1 signature identified herein as a mediator of reduced HIV-1 acquisition is further highlighted by our findings comparing ALVAC + Alum to ALVAC + MF59, the regimen used in HVTN702, as ALVAC + MF59 does not induce the same CREB1 signature seen in ALVAC + Alum, providing a potential mechanism for the lack of protection in HVTN702. Indeed, many of the key CREB1-driven immune pathways associated with reduced HIV-1 acquisition in RV144 were downregulated in the ALVAC + MF59 arm of P162. Together, our results have highlighted the need to change how correlates of vaccine efficacy are modeled and investigated. Single correlates fail to account for the multiplicity of cells that contribute to an effective immune response as well as the heterogeneity that exists across individuals in immune responses to HIV-1 that mediate protection from acquisition. Our integrated systems biology approach combining transcriptomics in multiple cohorts, ChIP–seq confirmation and in vitro mechanistic validation is required to robustly identify immune pathways that mechanistically reduce HIV-1 acquisition. Identification of adjuvants or vaccine components that activate these pathways is essential for developing an effective HIV-1 vaccine.
Methods
Study 36 vaccine trial design
Twenty-four Chinese-origin rhesus NHPs were used in the study. NHPs were housed at New England Regional Primate Research Center in Southborough, MA. This study was performed under Institutional Animal Care and Use Committee approval from Harvard Medical School (protocol HMS IACUC no. 03503). The animals were maintained in accordance with National Institutes of Health and Harvard Medical School guidelines.
The 24 NHPs were distributed in 5 groups with 5 animals in groups 1–4 and 4 animals in group 5. NHPs in group 1 (G1) were primed on weeks 0 and 4 with intramuscular immunizations with 5 × 107 plaque-forming units (pfu) of empty ALVAC (vCPpp, Sanofi-Pasteur), whereas NHPs in group 2 (G2) received intramuscular priming immunizations with 5 × 107 pfu of ALVAC-HIV-1 (vCP1521) at the same time. NHPs in group 1 were boosted with AIDSVAX B/E gp120 and 5 × 107 pfu of empty ALVAC administered intramuscularly on separate quadriceps muscles on weeks 12 and 23. Concurrently, NHPs in group 2 were boosted with AIDSVAX B/E gp120 and 5 × 107 pfu of ALVAC-HIV-1 administered the same way as in group 1. On weeks 12 and 23, NHPs in group 3 were immunized with same immunogens as group 1, and NHPs in group 4 were immunized with the same immunogens as group 2. Four NHPs in group 5 received only AIDSVAX B/E gp120 on weeks 12 and 23. NHPs in groups 3–5 received no priming immunizations. After resting for 30 weeks, on week 53 NHPs in groups 1–5 were boosted with the same immunogens as on weeks 12 and 23.
Vaccine immunogens
The ALVAC-HIV-1 and AIDSVAX B/E rgp120 immunogens used in this study are the same as those used in the RV144 Thai trial1. Both empty ALVAC control vCPpp and ALVAC-HIV-1 vCP1521 immunogens were received from Sanofi-Pasteur. ALVAC vCP1521 expresses subtype E (TH023 gp120) and subtype B (TM gp41) Env and subtype B Gag and Protease (LAI). AIDSVAX B/E gp120 that expresses HIV-1 subtype B (MN) and subtype E (A244) rgp120 were received from Global Solutions for Infectious Diseases.
HIV-1-specific binding antibody assay
Plasma HIV-1-specific antibodies were measured by a custom HIV-1 binding antibody multiplex assay as previously described49. Antibody titers (area under the curve) were determined by serial dilutions of rhesus plasma (1:80, 7-fold). All assays were run under Good Clinical Laboratory Practice–compliant conditions, including tracking of positive controls by Levey–Jennings charts using Title 21 Code of Federal Regulations Part 11–compliant software. Positive controls included a HIVIG and CH58 monoclonal antibody. Negative controls included in every assay were blank beads, HIV-1-negative sera and baseline (pre-vaccination) samples. To control for antigen performance, we used the preset criterion that the positive control titer (HIVIG and CH58) included on each assay had to be within ±3 standard deviations of the mean for each antigen (tracked with a Levey–Jennings plot with preset acceptance of titer (calculated with a four-parameter logistic equation, SigmaPlot, Systat Software)). Antibody measurements were acquired on a Bio-Plex instrument (Bio-Rad) using Title 21 Code of Federal Regulations Part 11–compliant software and the readout is in MFI. The preset assay criteria for sample reporting were: coefficient of variation per duplicate values for each sample was ≤15% and >100 beads counted per sample.
Sorting of NHP immune cell populations in Study 36
From anti-coagulated whole blood, DCs, CD4+ T cells and B cells were sorted following staining with monoclonal antibodies anti-CD45 V450 (clone DO58-1283; BD Pharmingen), anti-CD14 FITC (clone M5E2; BD Pharmingen), anti-HLA-DR PE (clone L243 (G46-6); BD Pharmingen), anti-CD20 ECD (clone B9E9; Beckman Coulter), anti-CD4 PerCP Cy5.5 (clone L200; BD Pharmingen), anti-CD123 APC (clone 7G3; BD Pharmingen), anti-CD11c APC (clone S-HCL-3; BD Pharmingen), anti-CD3 Alexa Fluor 700 (clone SP34.2; BD Pharmingen) and anti-CD8 APC.H7 (clone SK1; BD Pharmingen). After staining and washes, stained PBMCs were sorted using the BD FACSAria II. Doublets were excluded from analysis by gating singlets in forward scatter area versus forward scatter height and side scatter area versus side scatter height analysis. After gating on lymphocytes (CD45+ followed by forward scatter area versus side scatter area), total B lymphocytes were defined and sorted as CD3−CD8−CD20+ populations. DCs were gated and sorted as CD3−CD20−CD14−CD11c+/CD123+ cells. Sorted populations were collected into tubes containing RNA Protect (Qiagen) for RNA isolation.
RNA isolation, amplification and hybrization
Sorted cell populations were centrifuged at 800g for 10 min at room temperature, and 800 μl TRIzol reagent (Invitrogen) was added to lyse cells and dissociate nucleoprotein complexes. Following a phenol–chloroform extraction, the aqueous phase containing RNA was carefully collected. This aqueous RNA phase was then treated with ethanol; and finally, the RNA was suspended in RNase-free water. mRNA was amplified and biotinylated using the TargetAmp 2-Round Biotin-aRNA Amplification Kit 3.0 (EPICENTRE) for microarray studies. Hybridization was conducted using Human RefSeq-12 V4, according to Illumina’s direct hybridization protocol by the Genomics Analysis facility at Duke University Medical Center.
Microarray data preprocessing and analysis
Raw bead chip intensities were imputed using the k-nearest neighbors algorithm from the impute R package, and then those were quantile-normalized and log2 transformed. The LIMMA package was then used to fit linear regression models with the log2 gene expression as the dependent variable while the groups of interest (pre-vaccination versus post-vaccination time points; antibody responses) were independent variables to identify genes differentially expressed between conditions and found to be correlated with antibody response (IgG antibodies binding to HIV-1 Env V1V2 regions). A moderated t-test was used to assess the statistical significance of the association between gene expression and the groups of interest. Benjamini and Hochberg correction was applied to adjust for multiple testing.
Preranked GSEA was performed for each contrast and correlation against gene sets extracted from the ChEA (CHIP-X Enrichment Analysis) database20. ChEA is a database containing target gene sets for TFs that are generated by curating publically available datasets for known interaction of the TF with the promoter of the gene by ChIP–seq, or equivalent assay. The ChEA database was downloaded from the Harmonize portal. Gene sets found to be ALVAC induced in both the pre-vaccination and post-vaccination contrast and to be correlated with V1V2 titers were filtered as putative, differentially regulated TFs.
GSEA is an analytical method that tests for enrichment of high-ranking genes based on their correlation to an outcome (that is, differential expression after ALVAC, correlation to V1V2 titer) versus annotated pathways. This method possesses increased power to detect pathways because it is not reliant on strict P-value cutoffs to test for enrichment and ranks all genes (in this case, 30,948 unique genes) and then performs enrichment tests. GSEA was performed independently for each cell type, time point and outcome (that is, ALVAC induction and V1V2 correlation). Only TF gene sets that showed significant enrichment with a cell subset for all time points and outcomes were considered for further analysis in Fig. 1a. No compiled or common list of genes was used for GSEA analysis. This was done to account for temporal differences in TF-driven gene expression. Use of a common or compiled list would fail to account for the temporal diversity of TF responses that are critical for biological function.
Gene set variation analysis50 was then used to compute a sample-level gene set enrichment z-score at each experimental condition for the CREB1 signature, and this was correlated with differentially expressed genes with immune function Gene Ontology (GO) terms to assess the immune function regulated by differentially enriched transcriptional regulators. Genes recurrently and significantly associated with CREB1 signatures were used as inputs for the ClueGO addon in Cytoscape 3, to identify REACTOME and GO pathways (MSigDB) enriched among these genes by using a Fisher exact test with a P value of <0.05.
All code used for analysis is available at https://github.com/sekalylab/Study36.
Quantification of plasma cytokines using Meso Scale
A custom panel of cytokine and chemokines was quantified using the Meso Scale Discovery U-Plex platform (MSD). The panel was chosen based on cytokines implicated by transcriptional analysis. It is comprised of: eotaxin 3 (CCL26), FLT3LG, fractalkine (CX3CL1), GROα (CXCL1), I-TAC (CXCL11), IFN-α2a, IFN-χ, IL-12p40, IL-13, IL-18, IL-1α, IL-1β, IL-1Rn, IL-2, IL-6, IP10 (CXCL10), MCP1 (CCL2), TGF-β1, TGF-β2, TGF-β3, TNF, TPO and TRAIL.
Multivariate modeling
Regularized regression analysis was performed on Meso Scale data using the R package glmnet. Optimal lambda was determined using leave-one-out cross-validation: the model with the smallest mean square error for alpha values between 0 and 1 was selected for optimal predictive power, and coefficients for predictors were returned in their original scale. Standardized coefficients for comparison purposes were calculated using the Agresti method.
ChIP–seq
ChIP was performed on PBMCs from three NHP donors and three human donors. PBMCs were infected in vitro with a multiplicity of infection of 10:1 of ALVAC-HIV or empty ALVAC, with medium-cultured cells as a control. Cells were collected and crosslinked in 10% formaldehyde for 30 min. Cells were pelleted and stored dry at −20 °C for future work. Thawed pellets were sonicated using Bioruptor (Diagenode) to a fragment size of 300–500 base pairs (bp) and then incubated overnight with CREB1 antibody (clone 48H2, Cell Signaling Technology no. 9197). Antibody was pulled down, DNA–protein de-crosslinked and DNA purified/eluted using the commercial True MicroChIP kit (Diagenode). Libraries were prepared and indexed using the MicroPlex kit (Diagenode). Single-end reads of 100 bp in length were sequenced on the NovaSeq platform.
For bioinformatic analysis, human and NHP single-end fastq files were respectively aligned against the GRCh38 and Mmul_10 reference genomes using BWA, using the unstimulated medium condition as a control. Umapped and duplicated reads were filtered out using a combination of SAMtools, bedtools and Picard as described in the ENCODE ChIP–seq pipeline 2 (https://docs.google.com/document/d/1lG_Rd7fnYgRpSIqrIfuVlAz2dW1VaSQThzk836Db99c/edit#). High-quality reads from two donors per condition were used for analysis. Replicates were cross-correlated, and peaks were called using MACS2. Consistent peaks were selected using bedtools. Promoter peaks were associated with downstream genes on the basis on being found within the –3,000 bp to +200 bp region of a TSS, using the TxDb databases for GRCh38 and RheMac10 in combination with the R package ChIPseeker. Peaks were visualized using the Integrative Genomic Viewer on filtered bed files from annotated promoter peaks, using the fold change versus the unstimulated condition. Set overlap was tested and visualized using the R package SuperExactTest (https://www.nature.com/articles/srep16923).
cGAMP stimulation of primary human PBMCs and purified DCs and CD4+ T cells
cGAMP was obtained from Invivogen. Cryopreserved PBMCs from five healthy donors who underwent leukapheresis were thawed and rested in RPMI + 10% FBS + Pen/Strep + 10 mM HEPES overnight. Cells were stimulated for up to 60 min with 1 or 10 μM cGAMP. Cells were immediately collected, stained and analyzed for p-CREB1 using an LSRFortessa (BD). The staining panel included: anti-CD14 Percp eF710 (eBioscience, clone 61D3, catalog no. 46-0149-42), anti-CD16 AF700 (BD, clone 3G8, catalog no. 557920), anti-CD123 (BD, clone 7G3, catalog no. 564196), anti-CD11c (BD, clone B-ly6, catalog no. 560369), anti-CD19 V610 (Biolegend, clone SJ25C1, catalog no. 363024), anti-CD3 APC (BD, clone UCHT1, catalog no. 555335), anti-CD4 PECEF594 (BD, clone RPA-T4, catalog no. 562281), anti-CD8 BUV737 (BD, clone SK1, catalog no. 564629), anti-C45RA BV650 (Biolegend, clone HI1000, catalog no. 304136), anti-CCR7 PECY7 (BD, clone 3D12, catalog no. 557648), anti-CD27 APC-eFluor 780 (eBioscience, clone O323, catalog no. 47-0279-42), anti-CD95 BV711 (BD, clone DX2, catalog no. 563132), anti-CD25 BUV395 (BD, clone 2A3, catalog no. 564034), anti-FOXP3 FITC (Invitrogen, clone 236 A/E7, catalog no. 11-4777-42), anti-p-CREB PE (Cell Signaling, clone 87G3, catalog no. 14228S) and live/dead AmCyan (Life Technologies, catalog no. L34957). Immune subsets were analyzed for cGAMP-induced CREB1 phosphorylation by UMAP analysis and manual analysis of MFI using FLowJo (v10.5.3). Forskolin (Sigma) was used as a positive control for p-CREB induction.
For purification, pan-DC and total CD4+ kits (both from STEMCELL Technologies) were used to isolate populations for further analysis. Purified cells were stimulated with 10 µM cGAMP (Invivogen) and supernatants were collected after 24 h. Cytokine detection was performed using a custom Meso Scale kit (described above).
UMAP flow cytometry analysis
For bioinformatic analysis of flow cytometry data, a custom script was made using UMAP for dimensional reduction of data and visualization followed by RPhenograph analysis to generate clusters of cells based on their marker expression. This script has been uploaded to Github https://github.com/sekalylab/UMAP_Phenograph and is available to the public. Five thousand random events were selected from doublet-excluded, live cells from each sample for each analysis. This method allowed for visualization of high-dimensional data in two dimensions and for events to be clustered based on similar expression of flow cytometry markers, which led to identification of novel cell subsets.
Multiple correction
Adjustments for multiple comparisons were computed for all differential gene expression and pathway analyses in this study. Unless otherwise specified, reported P values are adjusted for multiple comparisons. In some cases (Extended Data Figs. 2a,b and 4c,d), a nominal P value threshold of 0.05 was used, and an adjusted P value of 0.25.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
No restrictions will be placed on data or material sharing used to generate Figs. 1–6. The microarray from Study 36 has been deposited in GEO using accession no. GSE180677. RV144 data have been deposited in GEO at GSE103733. ALVAC comp data have been deposited in GEO with the accession no. GSE181996. P162 data have been deposited in GEO with the accession no. GSE72624. ChIP–seq data have been deposited in GEO with the accession no. GSE180749. Raw flow cytometry data have not been deposited but will be shared upon request.
Code availability
All code utilized for microarray, ChIP–seq and UMAP analysis will be made available to the public at https://github.com/sekalylab/Study36 and https://github.com/sekalylab/UMAP_Phenograph.
References
Rerks-Ngarm, S. et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220 (2009).
Pitisuttithum, P. et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 194, 1661–1671 (2006).
Flynn, N. M. et al. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 191, 654–665 (2005).
Haynes, B. F. et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012).
Gottardo, R. et al. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS ONE 8, e75665 (2013).
Montefiori, D. C. et al. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J. Infect. Dis. 206, 431–441 (2012).
Tomaras, G. D. et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc. Natl Acad. Sci. USA 110, 9019–9024 (2013).
Rolland, M. et al. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 490, 417–420 (2012).
Pegu, P. et al. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIVmac251 acquisition in an immunization regimen that mimics the RV-144 Thai trial. J. Virol. 87, 1708–1719 (2013).
Vaccari, M. et al. Adjuvant-dependent innate and adaptive immune signatures of risk of SIVmac251 acquisition. Nat. Med. 22, 762–770 (2016).
Vaccari, M. et al. HIV vaccine candidate activation of hypoxia and the inflammasome in CD14+ monocytes is associated with a decreased risk of SIVmac251 acquisition. Nat. Med. 24, 847–856 (2018).
Gray, G. E. et al. Vaccine efficacy of ALVAC-HIV and bivalent subtype C gp120-MF59 in adults. N. Engl. J. Med. 384, 1089–1100 (2021).
Yu, Q. et al. Comparative analysis of tropism between canarypox (ALVAC) and vaccinia viruses reveals a more restricted and preferential tropism of ALVAC for human cells of the monocytic lineage. Vaccine 24, 6376–6391 (2006).
Harenberg, A., Guillaume, F., Ryan, E. J., Burdin, N. & Spada, F. Gene profiling analysis of ALVAC infected human monocyte derived dendritic cells. Vaccine 26, 5004–5013 (2008).
Mohan, T., Zhu, W., Wang, Y. & Wang, B. Z. Applications of chemokines as adjuvants for vaccine immunotherapy. Immunobiology 223, 477–485 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Bracci, L., La Sorsa, V., Belardelli, F. & Proietti, E. Type I interferons as vaccine adjuvants against infectious diseases and cancer. Expert Rev. Vaccines 7, 373–381 (2008).
Gaucher, D. et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 205, 3119–3131 (2008).
Querec, T. D. et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 10, 116–125 (2009).
Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010).
Zhu, C. Y. et al. Cell growth suppression by thanatos-associated protein 11(THAP11) is mediated by transcriptional downregulation of c-Myc. Cell Death Differ. 16, 395–405 (2009).
Horb, M. E. & Thomsen, G. H. Tbx5 is essential for heart development. Development 126, 1739–1751 (1999).
Offield, M. F. et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122, 983–995 (1996).
Baldin, V., Lukas, J., Marcote, M. J., Pagano, M. & Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 7, 812–821 (1993).
Delaloye, J. et al. Interleukin-1- and type I interferon-dependent enhanced immunogenicity of an NYVAC-HIV-1 Env-Gag-Pol-Nef vaccine vector with dual deletions of type I and type II interferon-binding proteins. J. Virol. 89, 3819–3832 (2015).
Roth, V. The generalized LASSO. IEEE Trans. Neural Netw. 15, 16–28 (2004).
Ancuta, P. et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J. Exp. Med. 197, 1701–1707 (2003).
Ogilvie, P., Paoletti, S., Clark-Lewis, I. & Uguccioni, M. Eotaxin-3 is a natural antagonist for CCR2 and exerts a repulsive effect on human monocytes. Blood 102, 789–794 (2003).
Waskow, C. et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 9, 676–683 (2008).
Xu, D. et al. IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur. J. Immunol. 30, 3147–3156 (2000).
Tassi, I. et al. The NF-κB regulator Bcl-3 governs dendritic cell antigen presentation functions in adaptive immunity. J. Immunol. 193, 4303–4311 (2014).
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Mesman, A. W. & Geijtenbeek, T. B. Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKε- and CYLD-dependent Bcl3 activation. Nat. Commun. 5, 3898 (2014).
Redmond, W. L., Ruby, C. E. & Weinberg, A. D. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit. Rev. Immunol. 29, 187–201 (2009).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Ahn, J., Gutman, D., Saijo, S. & Barber, G. N. STING manifests self DNA-dependent inflammatory disease. Proc. Natl Acad. Sci. USA 109, 19386–19391 (2012).
Sahoo, A. et al. Stat6 and c-Jun mediate Th2 cell-specific IL-24 gene expression. J. Immunol. 186, 4098–4109 (2011).
Tamada, K. et al. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J. Immunol. 164, 4105–4110 (2000).
Kearney, C. J., Randall, K. L. & Oliaro, J. DOCK8 regulates signal transduction events to control immunity. Cell Mol. Immunol. 14, 406–411 (2017).
Fulcher, J. A. et al. Galectin-1 co-clusters CD43/CD45 on dendritic cells and induces cell activation and migration through Syk and protein kinase C signaling. J. Biol. Chem. 284, 26860–26870 (2009).
Wang, M., Zhao, Y. & Zhang, B. Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923 (2015).
Guo, H. et al. NLRX1 sequesters STING to negatively regulate the interferon response, thereby facilitating the replication of HIV-1 and DNA viruses. Cell Host Microbe 19, 515–528 (2016).
Gao, D. et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 (2013).
Marcus, A. et al. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 49, 754–763 (2018).
Zaal, A. et al. Anaphylatoxin C5a regulates 6-sulfo-LacNAc dendritic cell function in human through crosstalk with Toll-like receptor-induced CREB signaling. Front. Immunol. 8, 818 (2017).
Moser, B., Wolf, M., Walz, A. & Loetscher, P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 25, 75–84 (2004).
Liu, Q. H. et al. Expression and a role of functionally coupled P2Y receptors in human dendritic cells. FEBS Lett. 445, 402–408 (1999).
Hara, S., Mizukami, H., Kuriiwa, F. & Mukai, T. cAMP production mediated through P2Y11-like receptors in rat striatum due to severe, but not moderate, carbon monoxide poisoning. Toxicology 288, 49–55 (2011).
Tomaras, G. D. et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 82, 12449–12463 (2008).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013).
Acknowledgements
We acknowledge A. Talla for generation of UMAP and Phenograph script for flow cytometry analysis; A. Trotch for assistance with running Meso Scale experiments; M. Vaccari for contribution of P162 data; the Yerkes NHP Genomics Core at Emory University for pooling and sequencing of ChIP–seq libraries; the veterinary staff who raised and cared for the animals used in Study 36 and P162; and the participants, clinical trial administrators and scientists who made RV144 possible. Study 36 was supported by Division of AIDS U19 grant AI067854 and UM1 grant AI100645 for the Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery (CHAVI-ID; B.H.); NIH, NIAID, Division of AIDS UM1 grant AI144371 for the Consortium for HIV/AIDS Vaccine Development (CHAVD; B.H.); the CCVIMC (OPP1032325) from the Bill and Melinda Gates Foundation (R.P.S. and R.K.); Harvard University Center for AIDS Research grant P30-AI060354; and New England Primate Research Center base grant OD011103. The Yerkes NHP Genomics Core is supported in part by NIH P51 OD011132 and sequencing data were acquired on an Illumina NovaSeq6000 funded by NIH S10 OD026799.
Author information
Authors and Affiliations
Contributions
J.A.T. designed and performed experiments, analyzed data, generated figures and wrote the manuscript. A.N.P. performed all bioinformatic analysis of data, generated figures and edited the manuscript. S.F. contributed to data analysis and manuscript preparation. M.B.L. helped perform the ChIP–seq experiment. A.S. contributed to revisions and manuscript preparation. G.F. provided samples and guidance for the P162 validation. G.T. contributed to Study 36 design and execution, performed antibody measurements and edited the manuscript. R.P.S. aided in experimental design and setup, data analysis and generation of figures, and wrote the manuscript. B.H., N.L. and S.S. designed and carried out Study 36, and B.H. and S.S. edited the manuscript A.G., K.F., K.C., K.S., M.L., N.Y. and R.P. assisted with Study 36. J.T. provided ALVAC for studies. P.W. identified the initial CREB signature in Study 36. M.L.R., N.L.M. and R.K. provided guidance on study design.
Corresponding authors
Ethics declarations
Competing interests
J.T. is an employee of Sanofi-Pasteur and provided the ALVAC vaccine components for all NHP and human studies. The other authors declare no competing interests.
Additional information
Peer review information Nature Immunology thanks Helder Nakaya and Shankar Subramaniam for their contribution to the peer review of this work. Peer reviewer reports are available. Zoltan Fehervari was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Study 36 design, vaccination schedule. sample collection strategy and bioinformatic analysis strategy.
a, NHP were immunized with ALVAC, empty insert [G1] or vCP1521 (HIV DNA encoding env, gag and pol)[G2], for two priming doses (Week 0 and 4) and two boosts along with recombinant Clade B/E gp120 (Week 12 and 23). PBMCs were isolated on Days 2/3 post Prime 1 (Week 0), 2 weeks post Prime 1 (Week 2) and 2 weeks post Boost 2 (Week 25) with DCs, CD4 + T cells and B cells purified by cell sorting. RNA was isolated from each cell type at each timepoint and microarray performed for transcriptional analysis. V1V2 IgG antibody titers were quantified at Week 14, Week 25, Week 53 and Week 55 post commencement of vaccine regimen. Additionally, plasma was isolated at the time of Prime 2 (Week 4) and 4 weeks post Boost 2 (Week 27). b, To identify TF target gene signatures which are associated with ALVAC immunogenicity, we implemented an analytical pipeline which generated independent GSEA analysis for each potential contrast (54 in total) in Study 36 in parallel and then identified those TF target gene signatures which were significant in all 3 cell types. First, we performed differential gene expression analysis of sorted DCs, CD4 + T cells and B cells by comparing post-vaccination timepoints (Week 0, Week 2 and Week 25) vs. pre-vaccination. GSEA using ChEA was performed on each of these 18 individual contrasts (3 cell types x 3 timepoints x 2 groups [G1,G2]) and this was termed ALVAC induced. Secondly, we correlated post-vaccination gene expression in the 3 sorted subsets (Week 0, Week 2 and Week 25) with IgG titers against V1V2 (Week 14, Week 25, Week 53 and Week 55). GSEA using ChEA was performed on each of these 36 individual contrasts (3 cell types x 3 microarray x 4 titers) and this was termed Correlate to V1V2. To identify TF target gene sets consistently ALVAC induced and correlating to V1V2, we intersect the 54 total GSEAs and highlighted TF target gene sets which were significant in all 3 cell types for ALVAC induction and correlation to V1V2.
Extended Data Fig. 2 IFN pathways are induced early in DCs and T cells but are not sustained during vaccination and do not correlate with V1V2 titers.
Upregulation of interferon signaling post-vaccination, compared to pre-vaccination, was determined for Group I and Group 2 in a, Dendritic cells and b, CD4+ T cells using preranked GSEA, without correction for multiple testing (p < 0.05). Induction of IFN pathways observed in both groups at Days 2/3 are absent by Week 25. In CD4 + T cells, only Group 2 shows upregulation of IFN and this response is only present at Days 2/3 suggesting that IFN upregulation post ALVAC vaccination is transient. N = 5 per group. A consolidated z-score generated for all IFN induced genes did not significantly correlate with V1V2 titers. Week 14 r = −0.7388, p = 0.154; Week 25 r = −0.3061, p = 0.6164; Week 55 r = −0.7993, p = 0.1046. c, To validate our GSEA findings in Study 36, we performed hypergeometric distribution testing. (Left) Significant enrichment of induced CREB1 target genes was observed for all 3 cell types at Weeks 0 and 2 in both G1 and G2. (Right) Significant enrichment of CREB1 target genes which correlate with V1V2 titers was found in all 3 cell types at Week 0 and Week 2 for the 4 titer timepoints measured. The lone exception was no significant enrichment for DCs at Week 0 to V1V2 titers at Week 53. These findings confirm our identification of CREB1 as ALVAC induced and a global correlate of V1V2.
Extended Data Fig. 3 ALVAC augments CREB1 driven gene expression by 24 hours post immunization and CREB1 signaling increases up to 72 hours post vaccination.
a, To more clearly understand the temporal nature of ALVAC induced CREB1, we accessed data from samples taken from the P162 study in which gene expression was quantified at 16, 24, 48 and 72 hrs after each of the 4 vaccination timepoints (n = 6 animals). GSEA using the CREB1 gene set from ChEA was performed to identify the temporal kinetics of ALVAC induced CREB1 by quantifying the fold induction of genes compared to pre-vaccination. In general, CREB1 target genes were suppressed at 16 hrs and induced at 24, 48 and 72 hrs. b, To determine if the same genes which were downregulated in some timepoints overlapped with those induced at others, a network was generated using Jaccard index to determine overlap in the CREB1 gene set leading edges for each of the timepoints. Red node color indicates induced, blue node color indicates suppressed. Node edge is colored per timepoint. Thickness of lines connecting nodes reflects the Jaccard index such that the thicker the line the more overlap. As can be seen, there is strong overlap by Jaccard index between the leading edges for the timepoints showing induced CREB1 target gene expression. By contrast, there is low overlap between blue nodes and red nodes indicating that the CREB1 target genes suppressed in the 4 timepoints are distinct from those induce at other timepoints.
Extended Data Fig. 4 CREB1 associated cytokine and chemokines in the plasma predict V1V2 titers and chemotaxis pathways are induced in Study 36.
a, Univariate correlations, using Pearson correlation, of Week 4 and Week 27 plasma cytokine levels to Week 53 and Week 55 V1V2 IgG. Statistical analysis performed using Pearson correlations (reported p.values. upper left: Eotaxin-3 = 0.0317, I-TAC = 0.017; upper right: Eotaxin-3 = 0.0457, IL-13 = 0.0199, IFNγ = 0.0094; lower right: GROα = 0.0187). b, Multivariate modeling using Lasso for Week 4 and Week 27 cytokines identifies combinatorial signatures of cytokines which strongly predict V1V2 titers. N = 5 animals from G2 arm of Study 36. c, In RV144, the individual participant z-scores of the cytokine/chemokine network from Fig. 2A was split into tertiles and associated with reduced HIV-1 acquisition using Log-rank test. Significantly reduced risk of HIV-1 acquisition was observed for the medium tertile (p = 0.005) along with substantial reduction in the high tertile (p = 0.10). d, GSEA analysis revealing ALVAC induced chemotactic signaling cascades in DCs, CD4 + T cell and B cells. e, GSEA analysis of GPCR signaling pathways in DCs and CD4 + T cells which correlate significantly with V1V2 titers. f, Subset specific distribution of cytokines and chemokines and their receptors which correlate, positively or negatively, with V1V2 titers in Study 36. Identifies common and unique cytokines/chemokines and their receptors among the 3 key immune subsets.
Extended Data Fig. 5 ChIP-seq confirms ALVAC induced CREB1 binding to immune genes in NHP and human PBMCs which overlap with transcriptional correlates of vaccine efficacy.
a, Venn diagram showing overlap between genes identified as enriched for CREB1 binding after ALVAC infection of NHP PBMCs and the ChEA CREB1 dataset. b, Bar plot showing significant overlap in genes identified by ChIP-seq as ALVAC induced in 2 or more of the infection conditions: empty ALVAC for 24 hrs, empty ALVAC for 48 hrs, ALVAC-HIV for 24 hrs and ALVAC-HIV for 48 hrs. c, Integrative Genomics Viewer (IGV) maps showing peaks of enriched CREB1 binding post ALVAC-HIV stimulation in the promoters of CX3CL1 (Fractalkine), CCL2 and Mamu-E, the NHP homolog of human HLA-E. d, Bar plot showing significant overlap between CREB1 genes identified as ALVAC induced, positive correlates of V1V2 in B cells from Study 36 and genes showing enriched CREB1 binding within 3 kB of their TSS in 1 or more of the ALVAC infection conditions by ChIP-seq. Significant overlap is observed for all combinations. e, In human PBMCs, enriched CREB1 binding was observed within 3kB of the TSS for the cytokine FLT3LG, antigen presentation gene HLA-DMB and TBK1 which is the kinase activated by STING. f, Venn diagram showing overlap between genes identified by ChIP-seq as enriched for CREB1 binding after 24 hr ALVAC infection of human PBMCs and ChEA CREB1 gene set. g, Bar plot of CREB1 target genes by ChIP-seq overlapped with the leading edge of CREB1 genes in RV144 which: 1) correlate with V1V2, 2) are induced post-vaccination or 3) are associated with reduced HIV-1 acquisition. Importantly, significant overlap is observed between CREB1 driven genes identified by ChIP-seq and CREB1 driven genes associated with reduced HIV-1 acquisition.
Extended Data Fig. 6 Genes and pathways reduced in MF59 are CREB1 driven and associated with reduced HIV acquisition in RV144.
a, GSEA analysis identifying immune related pathways which are significantly elevated in Alum compared to MF59 in P162. b, Network of the leading edge genes from pathways identified in Fig. 5d,e. Genes which are colored green are known CREB1 targets by ChEA and those in red are predicted CREB1 targets by promoter motifs. The majority of genes from these immune pathways are known or predicted targets of CREB1. c, The frequency of leading edge genes from the ALVAC induced CREB1 gene sets which correlate to V1V2 IgG titers was determined for all 3 cell subsets for all 12 contrasts (3 microarray timepoints compared to 4 V1V2 titers). The frequency of CREB1 driven genes which correlate to titers is significantly increased in all 3 cell subsets. This is true even for Week 25 microarray after antigen has been introduced into G1. Microarray timepoints (shapes): Circle = Week 0, Square = Week 2, Triangle = Week 25. V1V2 IgG titer timepoints: Black = Week 14, Red = Week 25, Blue = Week 53, Orange = Week 55. A two-sided Wilcoxon matched-paired signed rank test performed to compare frequency in G2 vs G1 paired at each individual contrast.
Extended Data Fig. 7 Manual FlowJo analysis confirms increased p-CREB + MFI and frequency in innate and adaptive immune cells.
a, p-CREB signal was quantified following treatment with Forskolin (n = 3), a known inducer of cAMP signaling, showing that p-CREB induction results in a shift in signal but is not bi-modal. b, Gating strategy for analysis. c, Histogram of p-CREB1 MFI in all cells and Clusters 5, 16, 17 and 20 showing higher p-CREB1 in these clusters compared to all cell. d, Density plots of UMAP analysis from Fig. 4 show that cGAMP induced p-CREB + populations are returned to basal levels after 60 minutes which is suggestive of true phospho-signaling. e, Representative histograms showing shift in p-CREB activity in total CD4 + T cells following 1 μM cGAMP stimulation for 15 m. f, Manual flow analysis of cGAMP treated samples (n = 5) in Fig. 4 confirms significant increases in p-CREB MFI for total CD4 + T cells, B cells, NK cells and other innate cells. Statistical analysis performed using a two-sided paired t-test. Line represents median and error bars are standard error of the mean.
Extended Data Fig. 8 Additional CREB1 related cytokines are induced in immune cells stimulated with cGAMP.
a, Median fold induction for all measured cytokines in purified DCs and CD4 + T cells treated with 10 uM cGAMP for 24 hours. b, TRAIL, GROα (CXCL1) and TGF-β1 which were all identified as positive correlates of V1V2 in Study 36 are also significantly induced by cGAMP treatment of purified DCs (black) and CD4 + T cells (blue) (n = 9), when these two groups are combined as distinct biological samples. Statistics performed using two-sided, paired t-test Lower line in violin plot is Q1, middle line is median and upper line is Q3. c, Proposed model of ALVAC induced CREB1 activation in mediating protective vaccine responses.
Supplementary information
Rights and permissions
About this article

Cite this article
Tomalka, J.A., Pelletier, A.N., Fourati, S. et al. The transcription factor CREB1 is a mechanistic driver of immunogenicity and reduced HIV-1 acquisition following ALVAC vaccination. Nat Immunol 22, 1294–1305 (2021). https://doi.org/10.1038/s41590-021-01026-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-021-01026-9
This article is cited by
-
Vaccine plus microbicide effective in preventing vaginal SIV transmission in macaques
Nature Microbiology (2023)
-
HIV vaccine candidate efficacy in female macaques mediated by cAMP-dependent efferocytosis and V2-specific ADCC
Nature Communications (2023)
-
Fighting the SARS-CoV-2 pandemic requires a global approach to understanding the heterogeneity of vaccine responses
Nature Immunology (2022)
-
Species-specific transcriptomic changes upon respiratory syncytial virus infection in cotton rats
Scientific Reports (2022)
