
Abstract
When helper T (TH) cell polarization was initially described three decades ago, the TH cell universe grew dramatically. New subsets were described based on their expression of few specific cytokines. Beyond TH1 and TH2 cells, this led to the coining of various TH17 and regulatory (Treg) cell subsets as well as TH22, TH25, follicular helper (TFH), TH3, TH5 and TH9 cells. High-dimensional single-cell analysis revealed that a categorization of TH cells into a single-cytokine-based nomenclature fails to capture the complexity and diversity of TH cells. Similar to the simple nomenclature used to describe innate lymphoid cells (ILCs), we propose that TH cell polarization should be categorized in terms of the help they provide to phagocytes (type 1), to B cells, eosinophils and mast cells (type 2) and to non-immune tissue cells, including the stroma and epithelium (type 3). Studying TH cells based on their helper function and the cells they help, rather than phenotypic features such as individual analyzed cytokines or transcription factors, better captures TH cell plasticity and conversion as well as the breadth of immune responses in vivo.
Similar content being viewed by others


Main
TH cell polarization is primarily geared toward responder cells that synergize, amplify and cooperate toward a distinct type of response, while repressing alternative responses at a certain time point of disease or infection. This is, to a large extent, achieved by a complex and tightly regulated network of activating and inhibiting cytokines. Aside from the cytokine pattern captured, helper properties are further expressed through surface molecules, pattern of migration and the ability to enter specific tissues. Here, we focus on what was traditionally used to define TH cells, namely the individual cytokines proposed to categorize TH cells. The expression of cytokines by TH cells depends on upstream signals from the encounter with antigen-presenting cells (APCs). This combination of cytokines lays, together with specific transcription factors (TFs) that control their expression, the foundation for the current classification of TH cell subsets. With the emergence of new technologies enabling us to simultaneously measure literally dozens of cytokines along with other markers such as TFs, integrins or chemokine receptors at the single-cell level1, it is no longer feasible to categorize TH cells based on a dominant cytokine or even a family of cytokines2. Also, by attempting to categorize every single TH cell based on individual cytokines or TFs, we may overlook the actual complex biology of differential responses and other involved cell types. Here, we focus on how the expanding TH cell universe can be reorganized based on the actual help provided toward the actual cellular targets, rather than on the momentary expression of certain cytokines and TFs.
Historical perspective
The categorization of T cells by their biological properties has provided us essentially with CD8+ cytotoxic killer and CD4+ TH cells. In 1971, an inverse relationship between humoral and cell-mediated immunity was observed by Chris Parish and Eddy Liew and others3, laying the foundation for TH cell bifurcation4,5,6. Eventually, Mosmann and Coffman described in 1986 that TH cells can be polarized to produce either interferon (IFN)-γ or interleukin (IL)-4, depending on their environment and stimulatory context7. Later, dominant TFs were found to drive this polarization program, namely T-bet for TH1 cells and GATA-3 for TH2 cells8,9,10. Importantly, one subset actively suppresses the other’s ability to produce its characteristic cytokines and TFs7.
Another, now well-established TH subset comprises Treg cells. Already in the early 1970s, experiments with thymectomized mice showed development of tissue damage in various organs, indicating the presence of a suppressive T cell subset developing in the thymus11,12. However, due to lack of reliable markers to distinguish these cells from other T cells, Treg cells underwent a history from being defined as Tr1 cells, when secreting the suppressive cytokine IL-10 in vitro, to being termed TH3 cells, when found to secrete transforming growth factor (TGF)-β upon induction of oral tolerance13,14. Nowadays, thymically hard-wired Treg cells are characterized by high expression levels of the high-affinity IL-2 receptor α-chain CD25 (ref. 15) and the TF FoxP3 (ref. 16) and are known to be of particular importance for maintaining immune homeostasis and preventing autoimmunity16.
Whereas the simple TH1–TH2 paradigm provided an easy explanation of immune responses toward intracellular and extracellular pathogens, respectively, numerous open questions emerged in the context of chronic inflammation and autoimmunity. The path for extending the TH family was cleared after it was noted that the IFN-γ-inducing cytokine IL-12 was not the critical factor for the induction of autoimmune pathology in preclinical models of chronic tissue inflammation, mimicking diseases such as multiple sclerosis (MS), rheumatoid arthritis and others. Instead, IL-23, which shares the p40 subunit with IL-12, was actually the main driver of the inflammatory response17,18,19. In addition to being pivotal for the development of pathogenic CD4+ T cells in neuroinflammation, IL-23 also triggers IL-17 expression20,21. Thus, it was recognized that TH1 cells were not the sole driving force for autoimmune pathology, at least in the context of experimental autoimmune encephalomyelitis (EAE), and the call was out for the identification of the true (pathogenic) TH cell subset(s) in this disease.
In 2005, IL-17-producing TH cells were described as a new entity22,23. This subset was readily accepted as an independent TH subset, probably due to its clear segregation from TH1 and TH2 cells, the induction of which seemed to antagonize the production of IL-17 (ref. 22). The definition of TGF-β and IL-6 as differentiation factors for these T cells in vitro24,25,26 and the identification of RAR-related orphan receptor γ (RORγt) as a critical TF for IL-17 secretion solidified the standing of an independent TH17 subset27. Even though the role of TH17 cells in tissue inflammation in general has been heavily debated, IL-17-producing cells have been clearly implicated in a number of chronic inflammatory diseases such as psoriasis, rheumatoid arthritis and Crohn’s disease (reviewed in ref. 28)29,30. Of note, neutralization of IL-17 in patients triggers fungal infection as a major frequent side effect, demonstrating the importance of IL-17 and IL-17-producing cells (such as TH17 cells) in anti-fungal control in mucosal tissues.
Already in 2000, another new subset was proposed, when two groups showed that B cell help in follicles was provided by specific TH cells that reside close to the B cell zone in secondary lymphoid structures31,32. These TH cells express the CXC chemokine receptor 5 (CXCR5) that is also expressed on mature B cells and were termed TFH cells. However, it was not until 2009 that BCL-6 was identified as the TF necessary for the generation of TFH cells33. Even then, the acceptance of TFH cells as an independent entity was strongly debated. This was partly due to the observation that the expression of canonical TH1, TH2 or TH17 cytokines such as IFN-γ, IL-4 and IL-17, respectively, was necessary to induce a proper class-switching reaction in B cells34,35,36. Although the regulation of the expression of these cytokines in TFH cells is not yet clear, it has been proposed that TFH cells differentiate independently of other TH subsets from naive CD4+ T cells when interacting with B cells upon initial activation by dendritic cells (DCs)37. Interestingly, the generation and retention of TFH cells depends on the same antagonistic TFs needed for germinal center B cell differentiation, namely BCL-6 and BLIMP1 (ref. 33), which may hint toward a role of specific niches as drivers for T cell diversity and plasticity.
The addition of new cytokines in the analysis workflow of immunology laboratories led to the description of additional TH subsets, such as TH9 (refs. 38,39), TH22 (refs. 40,41,42) and TH25 (ref. 43). To then adjust to this single-cytokine-based view on TH cells in immunity, even more subsets were coined. These include pathogenic versus non-pathogenic TH17, TH17.1, TH17.2 and TH5 cells, among others44,45,46. During this expanding discovery phase of new TH subsets, several voices warned against the idea that the identification of an individual cytokine expressed by TH cells should not automatically deliver a newly coined subset and that immunologists should keep an eye on the biology of these T cells and their role in immune responses47,48. The same holds true for the definition of a dominating TF needed to allow the ‘discovery’ of a new TH subset, especially as most of the subsequent findings were based on in vitro studies in which specific cytokine cocktails were applied to either naive or activated purified T cells.
Furthermore, the distinction of subsets requires not only ‘private’ master TFs but also, and maybe more importantly, stability and the ability to form memory. Stability is largely granted through epigenetic imprinting, which ensures the maintenance of the cells’ identity even after an extended period of time and without persistent antigenic threat. Even though there is some evidence that Treg cells can develop into TFH cells49 or intestinal intraepithelial cells50, genetic stability has been best described in Treg cells51. Some level of stability has been observed in TH1 and TH2 subsets52,53; however, not so much in TH17 cells54 or any of the other described subsets. At the present day, it is needless to say that the diversity of coined TH subsets has become exceedingly complex and also increasingly controversial among immunologists, as the designation of TH subsets beyond TH1, TH2 and TH17 cells remains debated.
Limitations of the current TH classification
The current TH subset classification reaches its meaningful limits when trying to categorize TH cells involved in the induction of pathologies. One prominent example is EAE, a preclinical model for the neuroinflammatory disease MS, in which the responsible TH subset was not fully elucidated despite decades of research (reviewed in ref. 55). For simplicity, we will here focus on tissue inflammation rather than immunity elicited by pathogens. As a frequently studied preclinical model for tissue inflammation, EAE was believed to be a TH1-mediated disease model because of the abundant IFN-γ-expressing TH cell infiltration in the central nervous system56,57. However, the observation that loss of IL-12 and IFN-γ signaling, respectively, led to EAE aggravation17,58,59 suggested that TH1 cells were not required for encephalitogenicity but may even have, at least partly, a protective role.
Shortly after, it was discovered that IL-23 signaling was pivotal for EAE induction and simultaneously was a potent inducer of numerous cytokines including IL-17 (ref. 21). This observation coincided with the claim that TH17 cells represent an independent TH cell subset22,23. This association in turn suggested that TH17 cells may represent the pathogenic, disease-initiating population in EAE. However, there are contradicting reports on the effect of canonical TH17 cytokines IL-17A and IL-17F on EAE. While one study described a milder course of EAE upon the depletion of IL-17A60, others failed to observe a tangible effect on the progression of EAE upon loss of IL-17A or IL-17F61, making conclusions on the involvement of TH17 cells in EAE more difficult. Only recently, it was shown that the effects of IL-17 on the disease course, aside from direct effects on the blood–brain barrier and perhaps astrocytes62,63, stem from its ability to shape the microbiome in the gut, thereby indirectly acting on central nervous system inflammation by shaping the systemic immune compartment64. The same study showed that exclusive IL-17 production by neuroantigen-specific T cells was dispensable for their pathogenic potential. Moreover, although the use of IL-17 fate-mapping mice showed that the use of complete Freund‘s adjuvant does favor the formation of IL-17-expressing TH cells, upon initiation of immunopathology, these cells showed a high degree of plasticity65. After tissue invasion, many of them produced high levels of IFN-γ, thereby raising the idea of an intermediate TH17–TH1 phenotype covering the ‘pathogenic’ TH cell subset.
An essential key-player cytokine of this pathogenic TH cell subset is the granulocyte–macrophage colony-stimulating factor (GM-CSF). In the context of EAE, GM-CSF is mainly produced by TH cells66 and has a dominant function in the development of the inflammatory cascade, as GM-CSF-deficient mice are completely resistant to EAE66,67,68. Furthermore, patients suffering from MS have elevated frequencies of GM-CSF-expressing TH cells69. It appears that GM-CSF, similar to tumor necrosis factor (TNF), can be transiently expressed by several TH subsets upon T cell receptor (TCR)-mediated stimulation (reviewed in ref. 70), making it difficult to allocate GM-CSF to one of the established TH subsets. Due to the inability to clearly define TH1 or TH17 cells as a pathogenic entity in EAE, a new GM-CSF-expressing TH subset was discussed66,71,72. This idea was supported by the observation that, while GM-CSF was clearly coexpressed with IFN-γ, coexpression with IL-17 was rarely observed73. However, GM-CSF expression was shown to be regulated by a complex transcriptional network downstream of the TCR, including the activity of TFs such as RORγt, NFAT, NF-κB, JNK–AP-1, PU.1 and BHLHE40 (reviewed in ref. 70); thus no individual dominant TF for GM-CSF expression has been identified so far. Regulation by different pathways might also indicate the need of tight control of GM-CSF expression to avoid accidental activation of this potent pro-inflammatory cytokine. Regardless of whether GM-CSF-expressing cells represent a new and independent cellular subset, the present categorization of TH cells is not able to unravel the bundle of distinct and overlapping TH subsets but rather limits the possibilities to define specific (disease-related) processes without colliding with the established nomenclature.
The power of plasticity
There is evidence that all TH cells, with the exception perhaps of Treg cells, retain a certain degree of plasticity upon differentiation into effector cells. This is a fortuitous feature, as it enables immune responses to adapt to changing circumstances based on incoming stimulating or inhibitory cues. Experiments regarding the stability of the single subsets showed that even fully differentiated TH1 and TH2 cells were able to switch their transcriptional signature when challenged under the respective conditions within the first 5 days of stimulation74,75. Prolonged stimulation, however, induced a more stabilized TH1 or TH2 program74. This indicates that polarized TH cells retain flexibility with regard to their transcriptional signature for several rounds of expansion, giving them enough time to adjust their response to stimulation. Especially TH17 cells have a particularly unstable lineage commitment, thus readily converting into TH1-like or Treg-like phenotypes (reviewed in ref. 76). The conversion of TH17 cells into TH1-like cells has especially been associated with the occurrence of organ-specific autoimmune diseases. Importantly, a high degree of TH flexibility cannot only be observed in laboratory animals under strictly defined experimental conditions but also in the human immune system. One example is the development of different vaccine-specific TH subsets that were not only diverse directly upon immunization but even able to change their ‘fate’ with following rounds of expansion77.
In sum, the flexibility of TH cells makes their classification based on cytokine patterns alone opaque and bulky. In a review article by O’Shea and Paul78, the authors acknowledged this challenge and proposed a continuum model in which TH cells are positioned across an orbital shape of states with the three TFs RORγt, T-bet and GATA-3 as the three extreme positions.
This ‘continuum model’ was certainly a step in the right direction, but, with increasing numbers of TFs and cytokines analyzed simultaneously, the anchor points of this orbital model extend into multidimensional space and can no longer help the visualization and conceptualization of T cell states. Therefore, we believe that the continual bifurcation of TH subsets no longer contributes to the understanding of the plasticity and functionality that these cells adduce but rather unnecessarily complicates our appreciation of dynamic immune responses. Current state-of-the-art methods such as single-cell RNA sequencing, assay for transposase-accessible chromatin with sequencing (ATAC-seq) and high-dimensional cytometry also failed to capture canonical polarized TH cells, particularly in vivo2,79. Instead, the data support the notion that TH cell-driven immune responses in mammals are highly diverse and complex. Kiner et al. recently also challenged the utility of TH archetypes in that unbiased analysis of intestinal TH cells shows that their phenotype is molded by the microbes they encounter79. This apparent breadth of TH cell states could be explained by the following: (1) TH cells are primed toward a certain lineage but then retain a high level of plasticity, or (2) TH cells are primed toward a diverse continuum, and there are no dedicated canonical lineages. Either way, dividing TH cells into increasing numbers of subsets, based on the cytokine production measured, may only apply to specific experimental conditions at a certain time point but does not contribute substantially to a better understanding of TH cell biology. Hence, we propose to take one step back and focus again on the actual helper function of TH cells and consider their polarization based on the target cells they ‘help’, akin to the designation of Treg and TFH cells, designations based on function rather than phenotype.
Reframing TH cell subsets
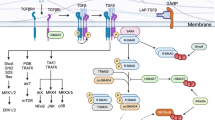
In 2018, Eberl and Pradeu proposed a unifying theory that takes the bigger physiological picture into account80. They started by picking up on the idea that the immune system is not activated by recognizing non-self per se but by the change in ‘normality’: the so called ‘discontinuity theory’81 (that builds upon the danger model that was proposed by Polly Matzinger in the 1990s82). The new theory considers three levels of immune responses: activation of the immune system by different means (for example, intracellular, tissular, extracellular), regulation of the immune response by cross-inhibition of different types of immune response (Fig. 1) and integration of the immune response into other vital processes necessary for maintaining homeostasis at the level of the whole organism80. The three types of responses that they described are loosely associated with the known concept of type 1, 2 and 3 immunity83. Accordingly, type 1 responses are induced by intracellular discontinuities, type 2 responses are involved in tissue-repair mechanisms to prevent entrance of pathogens, and type 3 responses are activated by discontinuities affecting extracellular space, such as fungi and bacteria in barrier tissues84. Such a simple classification would mirror that of other lymphocytes with helper function, namely ILCs (for review, see ref. 85).

From the perspective of a pathogenic insult, type 1 immune responses are typically triggered by intracellular pathogens. Multicellular organisms that cannot easily be phagocytosed induce type 2 responses that support the development of humoral immunity. Type 3 responses are initiated upon extracellular activation at barrier sites such as the skin, gut and other mucosal tissue. In this model, the three types of immune responses inhibit each other and are strengthened by auto-amplification.
We propose to extend this concept toward the initial definition of TH cells, namely their primary function: to provide help. TH cells are not predominantly killers or cleaners, but, as their name indicates, they support and enable other cells in the execution of their tasks. Depending on the context of activation, TH cells interact with different other cell types and produce a variety of cytokines, probably in varying concentrations and for a certain duration. This in turn acts on a palette of cell types including macrophages, DCs, monocytes, B cells or non-immune cell subsets that cross-regulate each other to achieve the desirable or adequate type of response. Therefore, we propose to consider TH cells by the type of responding cells that they target (Fig. 2). This classification, based on function rather than phenotype, is then further refined by the continuum model of O’Shea and Paul78 to acknowledge the plastic nature of TH cell states. However, while plasticity can be extensive, it is also limited by two major principles: first, cross-inhibitory interaction between type 1, type 2 and type 3 responses (as also suggested by Eberl80) and second, auto-amplification of established TH cell responses. Auto-amplification loops have been described for type 1, type 2 and type 3 responses, mostly based on T cell-derived cytokines that directly add back on their sources, re-enforcing their functional phenotype. IFN-γ86, IL-4 (ref. 87) and IL-21 (ref. 88) are examples of such autocrine feed-forward loop drivers for type 1, type 2 and type 3 responses, respectively.

TH cells can be classified by the primary target cells engaged. Type 1 responses target mononuclear phagocytes including macrophages and monocytes. The responding cells of type 2 immunity are predominantly mast cells, eosinophils and basophils, as well as B cells (particularly in germinal centers). Type 3 cytokines engage predominantly non-immune cells, such as epithelial cells across barrier tissues. In this model, the three types of immunity are interconnected, plastic and allow cross-talk when necessary. cDC, conventional dendritic cells.
Type 1 response
Type 1 responses are executed primarily by mononuclear myeloid cells, such as monocytes, macrophages and DCs. The most canonical type 1 cytokines produced by TH cells are IFN-γ and GM-CSF. Effects of IFN-γ in responder cells depend on the nature of the responding cell type89. The IFN-γ receptor (IFNGR) is a tetramer of two ligand-binding IFNGR1 chains and two signal-transducing IFNGR2 chains. While IFNGR1 is constitutively expressed on the surface of most cell types, IFNGR2 expression is more tightly regulated and predominantly found in phagocytes. More than 2,000 IFN-γ-responsive genes have been identified, including those encoding major histocompatibility complex (MHC)-I, MHC-II, nitric oxide synthase (NOS)2, various cell adhesion molecules such as vascular cell adhesion molecule (VCAM)1 and CD44, interferon regulatory factor (IRF)1–IRF9 and different tripartite motif (TRIM) genes90. IFN-γ is particularly important for APCs, as it not only induces the upregulation of MHC-I and MHC-II molecules but also slows lysosomal function in macrophages to enhance antigen processing91,92. Interestingly, other pro-inflammatory stimuli such as type I IFN, lipopolysaccharide and TNF can initiate a signaling cascade similar to that of IFN-γ93,94, thereby modulating the IFN-γ response but also possibly accounting for the mild phenotype of Ifng−/− and Ifngr−/− mice95. However, loss of IFN-γ signaling in mice leads to impaired clearance of several intracellular pathogens and a shift in the TH1–TH2 response (reviewed in (ref. 96)).
GM-CSF similarly acts as a potent communication conduit between T cells and myeloid cells97,98,99,100. The GM-CSF receptor is a heterodimer composed of the cytokine-specific α-chain and a β-chain that is shared with receptors for IL-3 and IL-5 (reviewed in ref. 101). Its cellular expression is even more restricted than the expression of the IFNGR, as the GM-CSF receptor is almost exclusively expressed by myeloid cells. In vitro stimulation with GM-CSF initiates the differentiation of DCs, granulocytes and macrophages, depending on the concentration of the cytokine102. The situation in vivo is more complex, although there is evidence that GM-CSF also has dose- and time-dependent effects in vivo103. In general, GM-CSF promotes survival, differentiation and activation of monocytes, macrophages and other phagocytes by engaging the Janus kinase (JAK)2–signal transducer and activator of transcription (STAT)5 and extracellular signal-regulated kinase (ERK) pathways104. Under certain inflammatory conditions, GM-CSF can be regarded as a pro-inflammatory mediator between TH cells and phagocytes (reviewed in ref. 105) and may also act on astrocytes to promote central nervous system pathology106,107. Hence, it is not surprising that GM-CSF-blocking antibodies are prominently used in clinical trials, for example, recently in the context of coronavirus disease 2019 (ref. 108).
Of note, among others, GM-CSF expression is induced by IL-23, which was also shown to be important for the modulation of ‘TH17’ responses20,66 and other type 3 immune responses (see below), making IL-23 both a type 1 and type 3 response-inducing cytokine, depending on circumstances (perhaps linked to its ability to signal through both STAT4 and STAT3). In this regard, it will be interesting to decipher additional factors causing a mainly destructive GM-CSF-driven type 1 response versus a protective IL-17-mediated type 3 response upon IL-23 exposure. Although it was argued that GM-CSF might serve as a marker for ‘destructive or pathogenic’ TH17 (or TH1–TH17, or TH17.1) cells, GM-CSF-producing cells preferably coexpress IFN-γ over IL-17 (refs. 66,69,71,73). Nevertheless, the relationship with IFN-γ appears to be a complex one, because both IFN-γ and its driver IL-12 effectively suppress GM-CSF production in T cells66. Of note, whereas T cells can sense IFN-γ, which has long been considered to aid in the maintenance of the TH1 phenotype, GM-CSF is not sensed by lymphocytes themselves. In spite of the apparent contradictions that emerge when TH cells are categorized by individual cytokine expression, the categorization of TH cells by the target cells that they help alleviates that problem and permits a better understanding of the actual biology of TH cells in type 1 immunity.
In sum, in type 1 responses, TH cells mainly target and activate phagocytic cells. While this communication aids in the elimination of intracellular pathogens, aberrant (dysregulated) type 1 responses, through persistent recruitment of phagocytes, can be drivers of immunopathology.
Type 2 response
Type 2 immune responses were initially described to primarily foster humoral immunity, and TH-derived type 2 cytokines predominantly help the B cell compartment and the involved intricacies to generate potent high-affinity antibodies. However, here again, the pure categorization of TH cell by their cytokine profile makes it much harder to capture the function of IL-4-secreting TH2 cells and TFH cells alike. As such, type 2 TH cells include not only TH2 and TFH cells but also TH1 cells, as all of them were shown to be necessary for humoral (type 2) immunity109,110. Typical type 2 cytokines are IL-4, IL-5 and IL-13. IL-4 was the first factor that was recognized to be crucial for B cell maturation and class switching, therefore recognizing TH2 cells as main providers of B cell help111. However, the deletion of TH2-associated genes did not cause loss of germinal centers. Later, it became apparent that IL-4 was solely needed for immunoglobulin (Ig)E class-switch recombination112 and that additional factors such as CD40 ligand (CD40L) and IL-21 were needed for fully functional B cell responses, which were attributed to TFH cells (reviewed in ref. 109). Of course, there are various flavors of TFH cells, which may warrant a TFH cell-specific nomenclature as suggested by Eisenbarth et al.113. Nevertheless, in this perspective, we consider their target, namely B cells, the reason why TFH cells are primarily type 2 TH cells.
Another important function of type 2 immunity beyond the engagement of B lymphocytes is the attraction and activation of eosinophils, mast cells and basophils during inflammatory responses. This is mainly achieved by the cytokines IL-5 and IL-13, which induce eosinophilia and goblet cell hyperplasia during helminth infections114. However, eosinophils, mast cells and basophils are not only type 2 effector cells, but they are also involved in the amplification of type 2 immunity by producing IL-4 and other type 2 mediators themselves. Eosinophil recruitment, for instance, can occur before infiltration of TH cells, which in turn stimulates APCs to initiate a type 2-promoting TH phenotype115,116. Although it is not fully understood which cell types induce the initial attraction of eosinophils, tissue-resident group 2 ILCs (ILC2 cells) might be involved, as they can react before the adaptive response is initiated117, making them important early-phase type 2 players. Furthermore, it was shown that the presence of ILC2 cells was required for a complete TH response, at least in the context of allergic inflammation118,119.
The alarmin IL-25, also known as IL-17E, was first reported to be secreted by TH2 cells and subsequently led to the coining of TH25 cells as an IL-25-producing entity that boosts type 2 responses by enhancing IL-4, IL-5 and IL-9 production via STAT5 activation120. Now we know that it can be produced by many different hematopoietic and non-hematopoietic cell types, such as mast cells, alveolar epithelial cells, brain capillary endothelial cells and others (reviewed in ref. 121). The exact mechanisms by which these cells induce and enhance type 2 responses are not fully understood yet; however, there is strong evidence that ILC2 cells act as type 2-response amplifiers122,123,124.
Another type 2 cytokine that has defined an independent TH subset is IL-9 (refs. 38,39). Initially believed to be a T cell growth factor125, IL-9 was soon recognized to be crucial for mast cell expansion and recruitment126. In this context, it is involved in the clearance of parasitic infections but may also play a role in promoting allergic inflammation (reviewed in ref. 127).
In sum, type 2 T cells, including TFH cells, primarily target B cells to aid in germinal center formation and class switch, whereas dysregulated type 2 immunity leads to allergic inflammation involving eosinophils, mast cells and basophils.
Type 3 response
Type 3 responses have been very well defined as barrier tissue-specific reactions to extracellular disturbances. Receptors for the critical cytokines IL-17 and IL-22 are expressed throughout the stromal and immune compartment, but dysregulated expression of these cytokines (IL-17A, IL-17F, IL-22, etc.) leads to dramatic immunopathology across barrier tissues (skin, lung, gut), with little to no signs of internal organ-specific effects61,128,129,130. Ectopic IL-17 expression has the most dramatic effect upon engagement of the IL-17 receptor complex in epithelial cells of the skin131. Apart from the production of antimicrobial peptides, IL-17-activated keratinocytes produce a set of chemokines and cytokines that in turn attract neutrophils into the skin (reviewed in ref. 132). Dysregulation of IL-17 in mammals also triggers psoriasiform inflammation, characterized by the cellular expansion of keratinocytes, and the influx of neutrophils. Targeting the type 3 immune response in patients suffering from psoriasis through neutralization of IL-17 or IL-23 dramatically alleviates clinical symptoms (reviewed in ref. 28). Strikingly, IL-23 is critical for both GM-CSF and IL-17 production in inflammatory conditions (as discussed above). This poses interesting questions about the regulation of IL-23 receptor signaling within different inflammatory conditions and cell types. In line with this, IL-23 was also shown to be released in response to nociceptor activation133,134, linking the immune system with the neuronal network. The notion that there is more to the immune system than simple host defense applies not only to type 3 immunity and pain sensation. A growing scientific field has attempted to decipher the interplay of the immune system and other physiological processes such as the neuronal network and the enteric system (reviewed in ref. 135).
In line with the notion that type 3 immune responses predominantly involve barrier tissues, physiological amounts of type 3 cytokines (such as IL-17A, IL-17F and IL-22) are involved in the control of mucosal pathogens, in particular, fungi (reviewed in ref. 136). However, IL-22-producing cells can be easily ‘reprogrammed’ into IFN-γ- or IL-4-expressing TH cells, illustrating one more time the dynamics of TH cell plasticity and indicating the importance of a flexible and collaborative environment for a functional immune system137.
Importantly, IL-17 and IL-22 production is readily observed in ILC3 cells and thymic educated γδ T cells, which are prominent and early responders in barrier tissue immunity, supporting the idea that a major portion of type 3 immunity is an evolutionary hard-wired mechanism of barrier protection138.
In summary, in contrast to type 1 and type 2 responses, type 3 responses are less targeted to distinct immune effector cells but activate and regulate non-immune cells. The code, which is used to induce type 3 responses (through, for example, IL-17 and the IL-20 family of cytokines), is likely phylogenetically old and is used by tissue-resident immune cells (such as ILC3 cells and γδ T cells) to communicate with their non-hematopoietic environment. Eventually, it has been co-opted by the adaptive immune system for host defense at lining tissues.
Summary and conclusion
The establishment of advanced single-cell analysis tools such as single-cell RNA-seq and high-dimensional cytometry revealed that the hitherto known classification of the TH cell universe, based on previously established cytokine patterns1,2,79, does not adequately capture the diversity and complexity of the mammalian immune system. For example, it was shown recently that TH cells isolated from the lamina propria could not be attributed to ‘classical’ TH1 or TH17 subsets but rather expressed a continuum of different (signature) cytokines79. Hence, we propose to take a step back to acknowledge the bigger picture, instead of focusing on small TH subsets that might simply represent an intermediate stage in their differentiation. By expanding the concept initially proposed by Eberl and Pradeu80 and integration of the until-now described subsets into a more comprehensive capture of immunity based on target cells of the TH response (Fig. 2), we propose the following nomenclature:
-
Type 1 TH cells that primarily activate and attract mononuclear phagocytes such as monocytes, macrophages and DCs
-
Type 2 TH cells targeting B cells and polymorphonucleated granulocytes such as mast cells, basophils and eosinophils
-
Type 3 TH cells acting on non-hematopoietic cells at barrier tissue sites, including epithelial cells and stromal cells.
This categorization is, in our opinion, superior to the coining of ever-new TH subsets and sub-subsets. We acknowledge that this concept is, however, also imperfect in that it does not capture all possible cellular states and their individual role in immune responses. Furthermore, we would hope to have solid molecular markers of TH cell states to better describe their biology. In lieu of such a ‘super-marker’ or molecular pattern of TH cell states, this simplified contextual ‘help’ framework proposed here is also not overly rigid. While polarized TH cells will in general fall into one of the three categories, this does not mean that their role in immunity is by any means inflexible. There is solid evidence of plasticity in memory TH cells and the ability to respond to different challenges with speed and agility. Hence, all attempts to categorize single TH cells observed during a snapshot within a complex immune response cannot truly give an account of the actual function and the role of individual TH cell in the development of a dynamic immune response. The physiological importance of TH differentiation must be the outcome of the response: the activation, attraction or modulation of responder cells. We hope that this perspective may help to establish a more intuitive classification of TH cell function, which will help to understand the growing complexity in this field. Lastly, this perspective here is not meant to cast a new nomenclature for TH cells but instead is meant to initiate the discussion to consider help function over phenotype as a potential stratifier for a more function-based categorization of TH cells.
References
Galli, E. et al. The end of omics? High dimensional single cell analysis in precision medicine. Eur. J. Immunol. 49, 212–220 (2019).
Tortola, L. et al. High-dimensional T helper cell profiling reveals a broad diversity of stably committed effector states and uncovers interlineage relationships. Immunity 53, 597–613 (2020).
Parish, C. R. & Liew, F. Y. Immune response to chemically modified flagellin: III. Enhanced cell-mediated immunity during high and low zone antibody tolerance to flagellin. J. Exp. Med. 135, 298–311 (1972).
Bottomly, K., Mathieson, B. J. & Mosier, D. E. Anti-idiotype induced regulation of helper cell function for the response to phosphorylcholine in adult BALB/c mice. J. Exp. Med. 148, 1216–1227 (1978).
Kappler, J. & Marrack, P. The role of H-2-linked genes in helper T-cell function. I. In vitro expression in B cells of immune response genes controlling helper T-cell activity. J. Exp. Med. 146, 1748–1764 (1977).
Tada, T., Takemori, T., Okumura, K., Nonaka, M. & Tokuhisa, T. Two distinct types of helper T cells involved in the secondary antibody response: independent and synergistic effects of Ia− and Ia+ helper T cells. J. Exp. Med. 147, 446–458 (1978).
Mosmann, T. R., Cherwinski, H., Bond, M. W., Giedlin, M. A. & Coffman, R. L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136, 2348–2357 (1986).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs TH1 lineage commitment. Cell 100, 655–669 (2000).
Zhang, D. H., Cohn, L., Ray, P., Bottomly, K. & Ray, A. Transcription factor GATA-3 is differentially expressed in murine TH1 and TH2 cells and controls TH2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272, 21597–21603 (1997).
Zheng, W. P. & Flavell, R. A. The transcription factor GATA-3 is necessary and sufficient for TH2 cytokine gene expression in CD4 T cells. Cell 89, 587–596 (1997).
Gershon, R. K. & Kondo, K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology 18, 723–737 (1970).
Nishizuka, Y. & Sakakura, T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166, 753–755 (1969).
Chen, Y., Kuchroo, V. K., Inobe, J. I., Hafler, D. A. & Weiner, H. L. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 265, 1237–1240 (1994).
Groux, H. et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389, 737–742 (1997).
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164 (1995).
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 (2003).
Becher, B., Durell, B. G. & Noelle, R. J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Invest. 110, 493–497 (2002).
Cua, D. J. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748 (2003).
Murphy, C. A. et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198, 1951–1957 (2003).
Aggarwal, S., Ghilardi, N., Xie, M. H., De Sauvage, F. J. & Gurney, A. L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278, 1910–1914 (2003).
Langrish, C. L. et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005).
Harrington, L. E. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 (2005).
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141 (2005).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
Mangan, P. R. et al. Transforming growth factor-β induces development of the TH17 lineage. Nature 441, 231–234 (2006).
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M. & Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006).
Ivanov, I. I. et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Zwicky, P., Unger, S. & Becher, B. Targeting interleukin-17 in chronic inflammatory disease: a clinical perspective. J. Exp. Med. 217, e20191123 (2020).
Borghi, M. et al. Antifungal TH immunity: growing up in family. Front. Immunol. 5, 506 (2014).
Bacher, P. et al. Human anti-fungal TH17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell 176, 1340–1355 (2019).
Breitfeld, D. et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 192, 1545–1551 (2000).
Schaerli, P. et al. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J. Exp. Med. 192, 1553–1562 (2000).
Johnston, R. J. et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009).
Mitsdoerffer, M. et al. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl Acad. Sci. USA 107, 14292–14297 (2010).
Olatunde, A. C., Hale, J. S. & Lamb, T. J. Cytokine-skewed TFH cells: functional consequences for B cell help. Trends Immunol. 42, 536–550 (2021).
Reinhardt, R. L., Liang, H. E. & Locksley, R. M. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 10, 385–393 (2009).
Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 29, 621–663 (2011).
Dardalhon, V. et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+IL-10+Foxp3− effector T cells. Nat. Immunol. 9, 1347–1355 (2008).
Veldhoen, M. et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 9, 1341–1346 (2008).
Duhen, T., Geiger, R., Jarrossay, D., Lanzavecchia, A. & Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 10, 857–863 (2009).
Trifari, S., Kaplan, C. D., Tran, E. H., Crellin, N. K. & Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat. Immunol. 10, 864–871 (2009).
Eyerich, S. et al. TH22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 119, 3573–3585 (2009).
Wu, L. et al. A novel IL-25 signaling pathway through STAT5. J. Immunol. 194, 4528–4534 (2015).
Cosmi, L. et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J. Allergy Clin. Immunol. 125, 222–230 (2010).
Ghoreschi, K. et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 467, 967–971 (2010).
van Hamburg, J. P. & Tas, S. W. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J. Autoimmun. 87, 69–81 (2018).
Zhou, L., Chong, M. M. W. & Littman, D. R. Plasticity of CD4+ T cell lineage differentiation. Immunity 30, 646–655 (2009).
Locksley, R. M. Nine lives: plasticity among T helper cell subsets. J. Exp. Med. 206, 1643–1646 (2009).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492 (2009).
Sujino, T. et al. Tissue adaptation of regulatory and intraepithelial CD4+ T cells controls gut inflammation. Science 352, 1581–1586 (2016).
Rubtsov, Y. P. et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010).
Avni, O. et al. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 3, 643–651 (2002).
Fields, P. E., Kim, S. T. & Flavell, R. A. Cutting edge: changes in histone acetylation at the IL-4 and IFN-γ loci accompany TH1/TH2 differentiation. J. Immunol. 169, 647–650 (2002).
Mukasa, R. et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32, 616–627 (2010).
Kunkl, M., Frascolla, S., Amormino, C., Volpe, E. & Tuosto, L. T helper cells: the modulators of inflammation in multiple sclerosis. Cells 9, 482 (2020).
Ando, D. G., Clayton, J., Kono, D., Urban, J. L. & Sercarz, E. E. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the TH-1 lymphokine subtype. Cell. Immunol. 124, 132–143 (1989).
Voskuhl, R. R. et al. T helper 1 (TH1) functional phenotype of human myelin basic protein-specific T lymphocytes. Autoimmunity 15, 137–143 (1993).
Ferber, I. A. et al. Mice with a disrupted IFN-γ gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 156, 5–7 (1996).
Krakowski, M. & Owens, T. Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 26, 1641–1646 (1996).
Komiyama, Y. et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol. 177, 566–573 (2006).
Haak, S. et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J. Clin. Invest. 119, 61–69 (2009).
Kebir, H. et al. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175 (2007).
Kang, Z. et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425 (2010).
Regen, T. et al. IL-17 controls central nervous system autoimmunity through the intestinal microbiome. Sci. Immunol. 6, eaaz6563 (2021).
Hirota, K. et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 (2011).
Komuczki, J. et al. Fate-mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-1β. Immunity 50, 1289–1304 (2019).
McQualter, J. L. et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J. Exp. Med. 194, 873–881 (2001).
Ponomarev, E. D. et al. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J. Immunol. 178, 39–48 (2007).
Galli, E. et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat. Med. 25, 1290–1300 (2019).
Sheng, W., Png, C. W., Reynolds, J. M. & Zhang, Y. T cell-derived GM-CSF, regulation of expression and function. Immunome Res. 11, https://doi.org/10.4172/1745-7580.1000098 (2015).
Herndler-Brandstetter, D. & Flavell, R. A. Producing GM-CSF: a unique T helper subset? Cell Res. 24, 1379–1380 (2014).
Sheng, W. et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 24, 1387–1402 (2014).
Noster, R. et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 6, 241ra80 (2014).
Murphy, E. et al. Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J. Exp. Med. 183, 901–913 (1996).
Hegazy, A. N. et al. Interferons direct TH2 cell reprogramming to generate a stable GATA-3+T-bet+ cell subset with combined TH2 and TH1 cell functions. Immunity 32, 116–128 (2010).
Lee, Y. K. et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009).
Becattini, S. et al. Functional heterogeneity of human memory CD4+ T cell clones primed by pathogens or vaccines. Science 347, 400–406 (2015).
O’Shea, J. & Paul, W. E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102 (2010).
Kiner, E. et al. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. 22, 216–228 (2021).
Eberl, G. & Pradeu, T. Towards a general theory of immunity? Trends Immunol. 39, 261–263 (2018).
Pradeu, T., Jaeger, S. & Vivier, E. The speed of change: towards a discontinuity theory of immunity? Nat. Rev. Immunol. 13, 764–769 (2013).
Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045 (1994).
Annunziato, F., Romagnani, C. & Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 135, 626–635 (2015).
Eberl, G. Immunity by equilibrium. Nat. Rev. Immunol. 16, 524–532 (2016).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Bradley, L. M., Dalton, D. K. & Croft, M. A direct role for IFN-γ in regulation of TH1 cell development. J. Immunol. 157, 1350–1358 (1996).
Kurtjones, E. A., Hamberg, S., Ohara, J., Paul, W. E. & Abbas, A. K. Heterogeneity of helper/inducer T lymphocytes: I. Lymphokine production and lymphokine responsiveness. J. Exp. Med. 166, 1774–1787 (1987).
Korn, T. et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature 448, 448–487 (2007).
de Veer, M. J. et al. Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69, 912–920 (2001).
Hertzog, P., Forster, S. & Samarajiwa, S. Systems biology of interferon responses. J. Interferon Cytokine Res. 31, 5–11 (2011).
Trost, M. et al. The phagosomal proteome in interferon-γ-activated macrophages. Immunity 30, 143–154 (2009).
Yates, R. M., Hermetter, A., Taylor, G. A. & Russell, D. G. Macrophage activation downregulates the degradative capacity of the phagosome. Traffic 8, 241–250 (2007).
Ahn, H. J. et al. A mechanism underlying synergy between IL-12 and IFN-γ-inducing factor in enhanced production of IFN-γ. J. Immunol. 159, 2125–2131 (1997).
Kovarik, P. et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-γ uses a different signaling pathway. Proc. Natl Acad. Sci. USA 96, 13956–13961 (1999).
Snapper, C. M. et al. Interferon-γ and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 236, 944–947 (1987).
Tau, G. & Rothman, P. Biologic functions of the IFN-γ receptors. Allergy 54, 1233–1251 (1999).
Croxford, A. L. et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43, 502–514 (2015).
Darrieutort-Laffite, C. et al. IL-1β and TNFα promote monocyte viability through the induction of GM-CSF expression by rheumatoid arthritis synovial fibroblasts. Mediators Inflamm. 2014, 241840 (2014).
Kobayashi, S. D. Spontaneous neutrophil apoptosis and regulation of cell survival by granulocyte macrophage-colony stimulating factor. J. Leukoc. Biol. 78, 1408–1418 (2005).
Zhan, Y. et al. The inflammatory cytokine, GM-CSF, alters the developmental outcome of murine dendritic cells. Eur. J. Immunol. 42, 2889–2900 (2012).
Barreda, D. R., Hanington, P. C. & Belosevic, M. Regulation of myeloid development and function by colony stimulating factors. Dev. Comp. Immunol. 28, 509–554 (2004).
Sun, L. et al. GM-CSF quantity has a selective effect on granulocytic vs. monocytic myeloid development and function. Front. Immunol. 9, 1922 (2018).
Guthridge, M. A. et al. Growth factor pleiotropy is controlled by a receptor Tyr/Ser motif that acts as a binary switch. EMBO J. 25, 479–485 (2006).
Guthridge, M. A. et al. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells 16, 301–313 (1998).
Becher, B., Tugues, S. & Greter, M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity 45, 963–973 (2016).
Sanmarco, L. M. et al. Gut-licensed IFNγ+ NK cells drive LAMP1+TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479 (2021).
Wheeler, M. A. et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020).
Mehta, P. et al. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: challenges and opportunities. Lancet Respir. Med. 8, 822–830 (2020).
Crotty, S. A brief history of T cell help to B cells. Nat. Rev. Immunol. 15, 185–189 (2015).
Smith, K. M. et al. TH1 and TH2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J. Immunol. 165, 3136–3144 (2000).
Howard, M. et al. Identification of a T cell-derived B cell growth factor distinct from interleukin 2. J. Exp. Med. 155, 914–923 (1982).
Kopf, M. et al. Immune responses of IL‐4, IL‐5, IL‐6 deficient mice. Immunol. Rev. 148, 45–69 (1995).
Eisenbarth, S. C. et al. CD4+ T cells that help B cells—a proposal for uniform nomenclature. Trends Immunol. 42, 658–669 (2021).
Koyasu, S. & Moro, K. Type 2 innate immune responses and the natural helper cell. Immunology 132, 475–481 (2011).
Shinkai, K., Mohrs, M. & Locksley, R. M. Helper T cells regulate type-2 innate immunity in vivo. Nature 420, 825–829 (2002).
Yang, D. et al. Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2–MyD88 signal pathway in dendritic cells and enhances TH2 immune responses. J. Exp. Med. 205, 79–90 (2008).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Halim, T. Y. F. et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 17, 57–64 (2016).
Halim, T. Y. F. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Fort, M. M. et al. IL-25 induces IL-4, IL-5, and IL-13 and TH2-associated pathologies in vivo. Immunity 15, 985–995 (2001).
Liu, Y., Shao, Z., Shangguan, G., Bie, Q. & Zhang, B. Biological properties and the role of IL-25 in disease pathogenesis. J. Immunol. Res. 2018, 6519465 (2018).
Moro, K. et al. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+ lymphoid cells. Nature 463, 540–544 (2010).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl Acad. Sci. USA 107, 11489–11494 (2010).
Renauld, J. C., Houssiau, F., Uyttenhove, C., Vink, A. & Van Snick, J. Interleukin-9: a T-cell growth factor with a potential oncogenic activity. Cancer Invest. 11, 635–640 (1993).
Townsend, M. J. et al. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity 13, 573–583 (2000).
Noelle, R. J. & Nowak, E. C. Cellular sources and immune functions of interleukin-9. Nat. Rev. Immunol. 10, 683–687 (2010).
Conti, H. R. et al. TH17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311 (2009).
O’Connor, W. et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 10, 603–609 (2009).
Sonnenberg, G. F. et al. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 207, 1293–1305 (2010).
Croxford, A. L. et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J. Invest. Dermatol. 134, 728–735 (2014).
Perera, G. K., Di Meglio, P. & Nestle, F. O. Psoriasis. Annu. Rev. Pathol. 7, 385–422 (2012).
Kashem, S. W. et al. Nociceptive sensory fibers drive interleukin-23 production from CD301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 43, 515–526 (2015).
Riol-Blanco, L. et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161 (2014).
Rankin, L. C. & Artis, D. Beyond host defense: emerging functions of the immune system in regulating complex tissue physiology. Cell 173, 554–567 (2018).
Sparber, F. & Leibundgut-Landmann, S. Interleukin-17 in antifungal immunity. Pathogens 8, 54 (2019).
Plank, M. W. et al. TH22 cells form a distinct TH lineage from TH17 cells in vitro with unique transcriptional properties and Tbet-dependent TH1 plasticity. J. Immunol. 198, 2182–2190 (2017).
Kubick, N. et al. Interleukins and interleukin receptors evolutionary history and origin in relation to CD4+ T cell evolution. Genes 12, 813 (2021).
Acknowledgements
We thank F. Sallusto and S. Sakaguchi for helpful comments on the manuscript. A.S.D. is supported by the Agence Nationale de la Recherche (ANR FoxoTic), the ARSEP foundation (ARSEP R19191BB) and the FRM (R20097BB); M.I. is supported by the European Research Council (ERC) Consolidator Grant 725038, the ERC Proof of Concept Grant 957502, the Italian Association for Cancer Research grants 19891 and 22737, the Italian Ministry of Health grants RF-2018-12365801 and COVID-2020-12371617, the Lombardy Foundation for Biomedical Research grant 2015-0010, the European Molecular Biology Organization Young Investigator Program and funded research agreements from Gilead Sciences, Takis Biotech, Toscana Life Sciences and Asher Bio. F.J.Q. is supported by NS102807, ES02530, ES029136 and AI126880 from the NIH; RG4111A1 from the National Multiple Sclerosis Society; and PA-1604-08459 from the International Progressive MS Alliance. F.G. is supported by the EMBO YIP, a BMRC Central Research Fund (UIBR) award and a Singapore NRF Senior Investigatorship (NRFI2017-02); TK is supported by the Deutsche Forschungsgemeinschaft (SFB1054, TRR128, TRR274, EXC 2145 (ID 390857198)) and the ERC (CoG647215); A.W. is supported by the Deutsche Forschungsgemeinschaft (TRR128, TRR156, CRC1292 and a Reinhard Koselleck prize); B.B. is supported by the Swiss National Science Foundation (733 310030_170320, 1030 310030_188450 and CRSII5_183478 to B.B.), H2020 Projects (826121, 847782) and the ERC (AdG 882424).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Immunology thanks R. Reinhardt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Zoltan Fehervari was the primary editor(s) on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tuzlak, S., Dejean, A.S., Iannacone, M. et al. Repositioning TH cell polarization from single cytokines to complex help. Nat Immunol 22, 1210–1217 (2021). https://doi.org/10.1038/s41590-021-01009-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-021-01009-w
This article is cited by
-
IFNγ drives neuroinflammation, demyelination, and neurodegeneration in a mouse model of multiple system atrophy
Acta Neuropathologica Communications (2024)
-
Strategies for targeting cytokines in inflammatory bowel disease
Nature Reviews Immunology (2024)
-
RORγt inverse agonist TF-S14 inhibits Th17 cytokines and prolongs skin allograft survival in sensitized mice
Communications Biology (2024)
-
The choroid plexus acts as an immune cell reservoir and brain entry site in experimental autoimmune encephalomyelitis
Fluids and Barriers of the CNS (2023)
-
An ocular Th1 immune response promotes corneal nerve damage independently of the development of corneal epitheliopathy
Journal of Neuroinflammation (2023)
