
Abstract
Influenza A, B and C viruses (IAV, IBV and ICV, respectively) circulate globally and infect humans, with IAV and IBV causing the most severe disease. CD8+ T cells confer cross-protection against IAV strains, however the responses of CD8+ T cells to IBV and ICV are understudied. We investigated the breadth of CD8+ T cell cross-recognition and provide evidence of CD8+ T cell cross-reactivity across IAV, IBV and ICV. We identified immunodominant CD8+ T cell epitopes from IBVs that were protective in mice and found memory CD8+ T cells directed against universal and influenza-virus-type-specific epitopes in the blood and lungs of healthy humans. Lung-derived CD8+ T cells displayed tissue-resident memory phenotypes. Notably, CD38+Ki67+CD8+ effector T cells directed against novel epitopes were readily detected in IAV- or IBV-infected pediatric and adult subjects. Our study introduces a new paradigm whereby CD8+ T cells confer unprecedented cross-reactivity across all influenza viruses, a key finding for the design of universal vaccines.
Similar content being viewed by others

Main
Although 2018 marked the 100th anniversary of the catastrophic Spanish influenza pandemic, influenza viruses remain a constant, global health threat. Three types of influenza viruses infect humans: type A (influenza A virus; IAV), type B (IBV) and type C (ICV). Two IAV subtypes (A/H3N2 and A/H1N1pdm09) and two IBV lineages (B/Yamagata/16/88-like and B/Victoria/2/87-like) co-circulate annually, causing seasonal epidemics of mild, severe or fatal respiratory disease, whereas ICV causes severe disease in children1,2,3,4,5. Antigenically novel IAVs generated by reassortment of the segmented genome and derived from animal reservoirs can also infect humans with high rates of morbidity and mortality1.
The search for a long-lasting, universal, broadly protective vaccine against influenza viruses is ongoing. Immune protection against influenza is mainly mediated by adaptive humoral and cellular responses1,2. Antibodies induced by seasonal inactivated influenza vaccine typically provide strain-specific immunity by targeting the variable head domain of the surface glycoprotein hemagglutinin. Although these antibodies can provide neutralizing immunity, the constant antigenic drift of hemagglutinin makes them poor targets for cross-protection. Broadly cross-reactive antibodies predominantly targeted to the conserved stem of hemagglutinin or at neuraminidase6 can provide heterosubtypic cross-reactivity across multiple IAV subtypes7 or across IBVs, but not heterotypic cross-reactivity across IAVs and IBVs, with the exception of one rare antibody clone (CR9114)8. Conversely, cytotoxic CD8+ T cells provide cross-protection across seasonal IAVs9,10 or IBVs11 and pandemic12,13,14,15 and avian16,17,18 IAVs by recognizing conserved virus-derived peptides presented by major histocompatibility complex class-1 (MHC-I) glycoproteins (human leukocyte antigens; HLAs) on the surface of infected cells. So far, 195 CD8+ T cell epitopes restricted by 24 different HLA allotypes have been identified for IAVs, 7 epitopes (restricted by HLA-A*02:01 or HLA-B*08:01) for IBV and no T cell epitopes for ICV (Immune Epitope Database; accessed 2 January 2018). After recognition of the peptide–MHC-I complex, CD8+ T cells kill virally infected cells and release antiviral cytokines (interferon-γ (IFN-γ) and tumor necrosis factor (TNF)). The breadth of CD8+ T cell cross-reactivity across antigenically novel viruses renders these cells promising targets for a universal vaccine. However, the current inactivated influenza vaccine formulation does not boost CD8+ T cells19. Thus, new vaccines are needed to harness the potential of cross-protective CD8+ T cells.
The establishment of universal immune memory against influenza viruses requires prior knowledge of conserved antigenic regions to facilitate immunogen design and assessment of immune responses. Although antibodies can be first isolated from serum and used to map the epitopes, identification of antigen-specific CD8+ T cells requires prior knowledge of the antigenic epitopes, including both peptides and restricting HLAs. This can inform the antigenic composition of T cell–based vaccines to focus the immune response towards conserved and protective epitopes. Here, we defined the CD8+ T cell cross-reactome against influenza A, B and C viruses, and identified novel IBV CD8+ T cells using immunopeptidomics20,21. We demonstrated that CD8+ T cells confer previously unrecognized, broad heterotypic cross-reactivity and characterized these responses in depth. Our data provide new insights into universal CD8+ T cells across IAVs, IBVs and ICVs, and suggest that combining universal CD8+ T cells with B cell–based vaccines might lead to a broadly protective influenza vaccine that does not require annual reformulation.
Results
Universal CD8+ T cell epitopes across IAV, IBV and ICV
To investigate the breadth of CD8+ T cell cross-reactivity across IAVs, IBVs and ICVs, we first assessed the conservation of previously identified IAV-specific CD8+ T cell epitopes across IAVs, IBVs and ICVs (Fig. 1a and Supplementary Fig. 1), as IAV-specific CD8+ T cells have been our the research focus so far. Our conservation analysis of >67,000 influenza segment sequences identified 31 conserved epitopes (with >70% amino acid identity) across IAV and IBV, and 8 epitopes across IAV, IBV and ICV types (Supplementary Table 1). Based on the prevalence of HLA-restricting molecules in the population and the nature of alterations within the peptide variants, we selected nine epitopes across both HLA-A (HLA-A*01:01, HLA-A*02:01 and HLA-A*03:01/A*11:01/*31:01/A*68:02) and HLA-B (HLA-B*07:02, HLA-B*44:02 and HLA-B*37:01) alleles (Table 1) for further investigation.

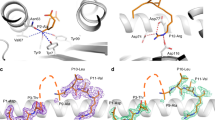
a, Conservation of known IAV epitopes in IBV and ICV. Bars indicate percentage conservation (average amino acid identity) of each peptide across the three types of viruses in the indicated number of sequences. IEDB, Immune Epitope Database. b, Immunogenicity of memory CD8+ T cells directed at conserved peptides in healthy adults. PBMCs were cultured with the peptides (as outlined in B) for ~10 d and responses were assessed in an IFN-γ ICS. Frequency of IFN-γ+CD8+ T cells after subtracting ‘no peptide’ control and responding donors are shown. Dots indicate individual donors (n = 3, except for A1-PB1591, n = 5, A2-PB1407, n = 4 and A2-PB1413, n = 6; different donors were assessed over at least two independent experiments), median and interquartile range (IQR) shown; ND, not detected. c, Conservation of PB1413–421 in orthomyxoviruses. Alignment of PB1 sequences derived from viruses representing each genus is shown. Box indicates the PB1413–421 peptide. d, A2-PB1413–421-mediated cross-reactivity across IAV, IBV and ICV. PBMCs were stimulated with one of the viruses for ~10 d and responses to the peptide were assessed in an ICS. A2-PB1413–421+CD8+ T cell responses measured directly ex vivo by ICS are shown for comparison. Representative concatenated FACS plots for IFN-γ production are shown. Data points indicate individual donors, median and IQR (n = 6 from two independent experiments). Statistical significance was determined using two-tailed Wilcoxon matched-pairs signed-rank test, *P < 0.05, **P < 0.005. e,f, B37-NP338–345-mediated (e) and A1-PB1591–599-mediated (f) cross-reactivity across IAV and IBV. On about day 10 of peptide culture, CD8+ T cell responses to either IAV or IBV variants were assessed by ICS. Dots indicate individual donors (n = 3 for B37-NP338 and n = 4 for A1-PB1591 from one experiment). d–f, ‘No peptide’ control was subtracted. d, *P = 0.049, **P = 0.0099.
To determine CD8+ T cell immunogenicity towards these epitopes, we probed memory CD8+ T cells within peripheral blood mononuclear cells (PBMCs) obtained from healthy adults using in vitro peptide expansion and measured IFN-γ after peptide re-stimulation. Three (A1-PB1591, n = 3; A2-PB1413, n = 5; B37-NP338, n = 3) of nine conserved CD8+ T cell epitopes recalled memory responses across multiple donors (Fig. 1b). These conserved CD8+ T cell peptides (PB1591–599, PB1413–421 and NP338–345) are restricted by three prominent HLA molecules (HLA-A*01:01, HLA-A*02:01 and HLA-B*37:01, respectively), providing broad global coverage as ~54% of the population carry at least one of these alleles.
The NMLSTVLGV PB1413–421 peptide in IAV (positioned as PB1414–422 in IBV and ICV; referred to as PB1413 hereafter) was universally (>98% of sequences) conserved (>99.9%) across IAV, IBV and ICV, but not in influenza D viruses, where a L7F alteration was found, or in other genera of the Orthomyxoviridae family (Fig. 1c). The PB1413–421 peptide has been reported as an IAV epitope22,23 shared in sequence with IBV24, however CD8+ T cell cross-reactivity has not been shown. To demonstrate the ability of HLA-A2-PB1413-specific CD8+ T cells to confer cross-reactivity across IAV, IBV and ICV, PBMCs obtained from HLA-A*02:01-expressing donors were stimulated in vitro with autologous PBMCs infected with IAV, IBV or ICV, followed by measurement of A2-PB1413+CD8+ T cells by IFN-γ on day 10 (n = 5) (Fig. 1d). In contrast to minimal IFN-γ production toward PB1413 directly ex vivo (Fig. 1d), ten-day culture with IAV-, IBV- or ICV-infected targets markedly increased the magnitude of A2-PB1413-specific CD8+ T cells (Fig. 1d), owing to expansion of A2-PB1413+CD8+ T cells towards all IAV, IBV and ICV. Our data thus provide evidence that memory A2-PB1413+CD8+ T cells are activated after stimulation with IAV-, IBV- or ICV-infected targets, suggesting that CD8+ T cells can exhibit universal cross-reactivity across IAV, IBV and ICV, and hence have a much broader cross-reactivity potential than has been previously thought.
Analysis of the remaining two conserved and immunogenic peptides (PB1591–599 and NP338–345 in IAV; BPB1590–598 and BNP394–401 in IBV) revealed variations at 1–2 amino acids (S2A and L8I for PB1591 and F1Y within NP338) between IAV and IBV, and a lack of conservation in ICV (Table 1). In vitro expansion with either IAV- or IBV-derived peptides showed unidirectional cross-reactivity, with IAV-expanded CD8+ T cells recognizing both IAV- and IBV-derived peptides. However, the IBV variants could not expand the number of CD8+ T cells directed at the cognate peptides, suggesting lower immunogenicity of these variants (Fig. 1e,f).
Collectively, our data demonstrate that human CD8+ T cells can confer heterotypic cross-reactivity across IAV, IBV and ICV. As the findings are based on the currently known IAV-derived epitopes and thus limited to IAV peptides, such universal cross-reactivity might be broader than defined here. Furthermore, our data suggest a need for identification of novel CD8+ T cell epitopes recognizing both IAV- and IBV-derived peptides that are restricted by a broad range of HLAs represented across different ethnicities.
Identification of novel HLA-A*02:01-restricted IBV epitopes
As there is a general lack of CD8+ T cell epitopes for clinically relevant and understudied IBVs, we identified novel CD8+ T cell epitopes derived from IBV viruses and presented by HLA-A*02:01, owing to the high global prevalence of this allele. We used immunopeptidomics to define peptides naturally processed and presented on the surface of IBV-infected cells. Epstein Barr virus-transformed B lymphoblastoid class I–reduced (C1R) cell lines stably expressing high levels of HLA-A*02:01 were used, together with parental C1R cells expressing background levels of HLA-B*35:01 and HLA-C*04:01 (ref. 25), to exclude peptides derived from these HLAs. Infection of C1R cells with the B/Malaysia strain resulted in high infection rates (~70% BNP+ cells) and cell viability (~93%) (Supplementary Fig. 2a). Liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of peptides isolated from HLA-A*02:01 revealed predominantly 9-mer (n = 1,490) peptides, followed by 11-mer (n = 695) and 10-mer (n = 589) peptides (Fig. 2a,b and Supplementary Fig. 2b). These peptides exhibited canonical anchor residues of HLA-A*02:01 ligands (leucine at P2 and leucine or valine at the C terminus26; Fig. 2b). Length distributions were similar for human peptides from uninfected cells and human or viral peptides from infected cells (Fig. 2c and Supplementary Fig. 2c). Analyses from two experiments yielded 73 potential HLA-A*02:01-presented IBV-derived peptides, with ~64% overlap between experiments (Supplementary Table 2). The IBV-derived peptides originated from hemagglutinin (BHA) (22.3%), followed by BNP (16.4%) and BM1 (11.9%), with all IBV proteins contributing to the HLA-A*02:01 immunopeptidome, except BM2 and NB (Fig. 2d). In contrast, peptides from BM2 were identified with high confidence as HLA-II ligands (Supplementary Fig. 2d and Supplementary Table 2). Of 73 HLA-A2-binding IBV peptides, 67 were synthesized for further investigation.

a, Immunopeptidomics outline. b, Peptide-binding motifs for host and IBV HLA-A*02:01 ligands generated from combined nonredundant lists of 9-mer, 10-mer and 11-mer, using Icelogo by the static reference method against the SWISS-PROT human proteome. c, Length distribution of filtered HLA-A*02:01 ligands (nonredundant by sequence) from uninfected (single experiment) and B/Malaysia-infected (two independent experiments) C1R.A*02:01 cells. Numbers of peptides of each length identified from the human proteome (5% FDR cutoff) and B/Malaysia proteome (all confidences) are shown. d, Distribution of IBV-derived HLA ligands (nonredundant by sequence) across the B/Malaysia proteome identified as probable HLA-A*02:01 ligands. Pooled data from two independent experiments. e–g, In vitro screening of novel peptides in human HLA-A*02:01-expressing PBMCs. e, Representative concatenated FACS plots for each peptide pool are shown from one donor, with a representative mock (unstimulated) control of a day 9–10 T cell line outlined for comparison. Frequency of IFN-γ+TNF+CD8+ T cells for each pool. Dots indicate individual donors; median and IQR are shown (n = 11, from at least two independent experiments). f, Frequency of responding donors for each pool (n = 11). g, Frequency of IFN-γ+TNF+CD8+ T cells directed toward individual peptides from pool 2 (n = 6 from two independent experiments); median and IQR are shown. h, Representative FACS plots for a positive CD8+ T cell response directed toward each peptide. Frequency of IFN-γ+TNF+CD8+ T cells. Donors are color coded; medians and IQRs are shown (n = 6 from one experiment). i,j, Immunodominance of universal CD8+ T cells during in vitro IAV or IBV infection. Responses during IAV infection against A2-M158, A2-PA46 and A2-PB1413 (i) and during IBV infection against A2-BHA543, A2-BNS1266 and A2-PB1413 (j). Bar charts show the contribution of each peptide to the total measured (sum of tetramer+ (Tet+)) response. n = 11 from one experiment. **P = 0.002, ***P = 0.001 (i); *P = 0.0352, #P = 0.0312, **P = 0.0098 (j).
Novel IBV BHA543 + and BNS1266 +CD8+ T cell epitopes in humans
To dissect IBV-specific CD8+ T cells toward the 67 LC–MS/MS-identified IBV peptides, we probed memory CD8+ T cell pools in HLA-A*02:01-expressing individuals. We assigned peptides to six pools of 10–12 peptides, avoiding overlapping peptides in the same pool. We established CD8+ T cell lines for each of the six peptide pools, and then re-stimulated each T cell line with the cognate pool in an IFN-γ and ΤΝF intracellular cytokine staining (ICS) assay (Fig. 2e,f). CD8+ T cell responses were predominantly targeted towards pool 2 (80% of donors, n = 11), with smaller responses detected for pools 1, 3, 4 and 6 (Fig. 2e and Supplementary Fig. 3a). Dissection of pool 2 identified A2-BHA543–551 as the prominent epitope among HLA-A*02:01+ donors (n = 6) (Fig. 2g). Smaller responses towards A2-BHA538–551, A2-NS1266–274, A2-BNS1264–274 and BM1132–140 were detected in some donors (Supplementary Fig. 3b). To validate these responses independently, we established CD8+ T cell lines towards individual immunogenic peptides. CD8+ T cell responses to A2-BHA543–551 were of the greatest magnitude (median 7.35%; n = 6) and more frequent among donors (6 of 6) than A2-NS1266–274 and A2-BM1132–140 (0.035% and 0.025%, respectively) (Fig. 2h and Supplementary Fig. 3b). Thus, our analysis identified five novel peptides recognized by CD8+ T cells in complex with HLA-A*02:01, with BHA543–551 being most prominent.
Having identified novel IBV CD8+ T cell epitopes, we determined the conservation of the most prominent peptides, BHA543–551 and BNS1266–274, across IBVs. Both peptides were highly conserved at >98% in >14,000 sequences per segment, spanning both lineages and 77 years (1940–2017) (Supplementary Fig. 3c). Although peptides identified by immunopeptidomics were highly conserved (>70%) in IAV (n = 6 peptides) or in ICV (n = 1) (Supplementary Fig. 3d), these were not immunogenic.
Overall, immunopeptidomics identified 73 novel IBV-derived HLA-A*02:01 peptide ligands. We tested 67 for immunogenicity; CD8+ T cell responses were targeted predominantly to BHA543–551, and highly conserved across IBV, but not IAV or ICV.
Universal PB1413 +CD8+ dominate over BHA543 +CD8+ T cells in IBV infection
Our data described so far identified three conserved HLA-A*02:01-restricted epitopes for IBV: the universal A2-PB1413 and two IBV-specific epitopes (A2-BHA543–551 and A2-NS1266–274, hereafter called A2-BHA543 and A2-BNS1266). To understand the immunodominance hierarchy of the universal A2-PB1413+CD8+ T cells after IAV or IBV infection, we established IAV- or IBV-specific CD8+ T cell lines in vitro from PBMCs of healthy adults (n = 11) and assessed tetramer-specific CD8+ T cells against IAV epitopes (A2-M158–66 (A2-M158), A2-PA46–54 (A2-PA64) and A2-PB1413) and IBV epitopes (A2-BHA543, A2-BNS1266 and A2-PB1413). Consistent with IFN-γ staining (Fig. 1d), A2-PB1413-tetramer detected universal A2-PB1413+CD8+ T cells within both IAV- or IBV-specific CD8+ T cell lines, although these cells displayed differential immunodominance hierarchies after either IAV or IBV infection (Fig. 2i,j). Within IAV-specific CD8+ T cell lines, A2-M158-tetramer+CD8+ T cells were significantly dominant (median 3.9% tetramer+CD8+ T cells; detected in all 11 donors) over universal A2-PB1413+ (0.12%; detected in 10 of 11 donors) and subdominant A2-PA46+CD8+ T cells (0.05%) (Fig. 2i). Conversely, the universal A2-PB1413 epitope within IBV-specific lines was immunodominant (0.3%; detected in 8 of 11 donors) over the IBV-specific A2-BHA543 (0.11%; detected in 10 of 11 donors) and A2-BNS1266 epitopes (0.01%) (Fig. 2j). These data demonstrate that (1) the universal A2-PB1413 and the newly identified IBV-specific A2-BHA543 and A2-BNS1266 CD8+ T cells can be expanded in number after virus stimulation in vitro, and (2) immunodominance of the universal A2-PB1413 epitope depends on influenza type.
Universal PB1413 +CD8+ T cells recruited during human IAV and IBV infection
To evaluate the recruitment and activation of universal A2-PB1413–421+CD8+ T cells during influenza virus infection, we analyzed PBMCs from three clinical cohorts of PCR-confirmed IAV- or IBV-infected pediatric and adult individuals (Fig. 3). Using tetramer-associated magnetic enrichment (TAME), we detected influenza-specific CD8+ T cells directly ex vivo in IAV- and IBV-infected pediatric and adult subjects (Fig. 3a,b)27,28,29. A healthy adult cohort and HLA-A*02:01-positive subjects hospitalized with a noninfluenza respiratory illness (influenza-PCR negative) were analyzed for comparison (Fig. 3b). A2-M158- and A2-PB1413-specific CD8+ T cells were detected in 100% and 50% of IAV+ individuals (n = 16), respectively, whereas A2-BHA543- and A2-PB1413-specific CD8+ T cells were detected in 75% and 87.5% of IBV+ individuals, respectively (n = 8). The frequency of A2-M158- and A2-PB1413-specific CD8+ T cells in the blood was significantly increased (4.3- and 6-fold increase, respectively) in IAV-infected subjects as compared to memory CD8+ T cells in healthy donors (Fig. 3c). The numbers of A2-BHA543- and A2-PB1413-specific CD8+ T cells in IBV-infected subjects increased 2.2- and 2.6-fold, respectively, above the healthy donors, however these increases did not reach statistical significance, most probably owing to the differential age distribution in IBV-infected, but not IAV-infected, subjects compared to healthy controls. In influenza-negative hospitalized subjects, CD8+ T cells for three specificities were in the same range as for the healthy donors (Fig. 3c). Notably, tetramer+CD8+ T cells for all specificities were detected across all age groups (Fig. 3d).

a–d, Tetramer-specific CD8+ T cells in healthy and influenza-infected individuals. a, Ex vivo TAME on PBMCs from healthy and infected donors. Representative FACS plots are shown. b, Characteristics of healthy and influenza-infected or influenza-negative influenza-like illness (ILI) cohorts used in this study. c, Precursor frequency of tetramer+ cells in healthy controls (HC), influenza-infected individuals and influenza-negative ILI subjects (n = 6 for flu-negative (flu-neg.) ILI, n = 8 for IBV and n = 24 for IAV, assessed over at least two independent experiments). Data points for flu-negative ILI were pooled across CD8+ T cell specificities owing to their varying detection levels across the six donors (5 of 6 for A2-M158, 2 of 6 for A2-PB1413 and 1 of 6 for A2-BHA543). Statistical significance was determined using a two-tailed Mann-Whitney test, *P = 0.03, **P = 0.0037. Median and IQR are shown. d, Precursor frequency of tetramer+ CD8+ T cells in healthy and influenza-infected individuals across age. e, Expression profiles of tetramer+CD8+ T cells for activation markers CD38 and Ki-67. Representative FACS plots are shown. Frequency of CD38+Ki-67+ tetramer+ CD8+ T cells from healthy controls (n = 3–5) and influenza-infected donors (n = 6 for flu-negative ILI, n = 8 for IBV and n = 24 for IAV, assessed over at least two independent experiments). Statistical significance for changes in the frequency of CD38–Ki-67– cells was determined using a two-tailed Mann-Whitney test A, *P = 0.017, **P = 0.0025, ##P = 0.0095.
Tetramer-positive A2-PB1413+CD8+, IBV-A2-BHA543+ and IAV-A2-M158+ CD8+ T cells detected in IAV- or IBV-infected subjects displayed increased CD38+ and Ki-67+ expression (Fig. 3e and Supplementary Fig. 4), representing an activated phenotype during human viral infections30,31,32. This suggests that they are recruited during influenza virus infection. This was not observed in the influenza-negative cohort, indicating that this activation is influenza-specific. Upregulation of additional activation markers, HLA-DR and PD-1, was also increased on tetramer+CD8+ T cells (Supplementary Fig. 5). The observed variability in numbers and phenotype is probably due to (1) the donors’ age range and exposure history and (2) varying times of sampling after influenza virus infection (Fig. 3b). Indeed, CD8+ T cell responses after human influenza A pandemic H1N1 infection peak within 7 d and then contract rapidly33. Additionally, the magnitude and activation status of peripheral CD8+ T cells can underrepresent virus-specific cells at the site of respiratory infections31.
These data show that A2-PB1413+CD8+ T cells are universal and can be detected with an activated phenotype in HLA-A*02:01-expressing influenza-infected subjects following either IAV or IBV. Additionally, activated CD8+ T cells specific for A2-BHA543–551, the epitope identified by immunopeptidomics, can be detected during IBV infection, illustrating the ability of mass spectrometry to identify novel peptide ligands.
Tissue-resident memory universal PB1413 +CD8+ T cells in human lungs
As human memory CD8+ T cells also reside outside the blood circulation34,35,36, we used rare human lung samples from deceased HLA-A*02:01-expressing organ donors (n = 5) to assess the presence of universal A2-PB1413+CD8+ T cells at the site of infection. We also used human spleens (n = 11), tonsils (n = 4) and lymph nodes (n = 4) to assess the presence of influenza-specific CD8+ T cells in the secondary lymphoid organs (SLOs), where memory CD8+ T cells are enriched. CD8+ T cells specific for A2-M158 (4 of 5), A2-PB1413 (2 of 5), and A2-BHA543 (1 of 5) were detected within human lung (Fig. 4a). Similarly, CD8+ T cells directed at A2-M158 (17 of 19), A2-PB1413 (6 of 19) and A2-BHA543 (4 of 19) were detected within human SLOs. Importantly, the majority of A2-PB1413+ and A2-BHA543 CD8+ T cells exhibited a tissue-resident memory (TRM) CD69+CD103+CD45RO+ phenotype in human lung but not in SLOs (Fig. 4b), with central (CD27+CD45RA–) or effector (CD27–CD45RA–) memory-like phenotype dominating in SLOs (Fig. 4c). This indicates the presence of universal A2-PB1413+ tissue-resident memory CD8+ T cells in human lung and memory pools in human SLOs.

a, Ex vivo detection of universal CD8+ T cells in human lung and secondary lymphoid organ (SLO; spleen, tonsils and lymph nodes) samples. Frequency of tetramer+CD8+ T cells (n = 8, lungs; n = 11, spleens; n = 4, tonsils; n = 4, lymph nodes); bars indicate the median. b, Phenotype of tetramer+CD8+ T cells was based on CD103 and CD69 expression within CD45RO+tetramer+CD8+ T cells. c, Phenotype of tetramer+ CD8+ T cells based on CD27 and CD45RA expression. Representative FACS plots are shown. Mean and s.e.m. are shown.
Overall, effector and memory IAV-A2-M158+, IBV-A2-BHA543+ and universal A2-PB1413+ CD8+ T cells can be detected directly ex vivo in blood and SLOs of healthy individuals, and tissue-resident IAV-A2-M158-specific and universal A2-PB1413-specific memory CD8+ T cells can be detected in human lung.
Single-cell RNA analysis of universal and IBV CD8+ T cells
To further understand the recruitment and activation of universal and novel IBV-specific CD8+ T cells during influenza virus infection, we used single-cell RNA sequencing (scRNA-seq) to assess the transcriptome of ex vivo–isolated tetramer+CD8+ T cells from longitudinal PBMC samples obtained from an IBV-infected HLA-A*02:01-expressing individual. Infection with a B/Victoria strain was confirmed by PCR37 and serological analysis (Supplementary Fig. 6a,b). Blood samples were obtained at baseline (~3 months before infection), and on 14 days, 3 months and 1.5 years after IBV infection (Fig. 5a). A2-PB1413+CD8+ T cells were detected at baseline at 19 tetramer+ per 106 CD8+ T cells, then increased 19-fold to 367 tetramer+ per 106 CD8+ T cells on day 14 and remained at a similar level (327 tetramer+ per 106 CD8+ T cells) for up to 1.5 years after infection (Fig. 5b). Conversely, A2-BHA543+CD8+ T cells were undetectable at baseline, suggesting that this was the first IBV infection for this donor, despite a previous immunization against the B/Yamagata strain with inactivated vaccine not eliciting CD8+ T cells19. A2-BHA543+CD8+ T cells increased to 73.4 tetramer+ per 106 CD8+ T cells on day 14 after infection; this was five-fold lower than for universal A2-PB1413+CD8+ T cells, and close to the detection level at 1.5 years. Thus, A2-PB1413+CD8+ T cells were assessed at all time points, whereas IBV-specific A2-BHA543+CD8+ T cells were analyzed on day 14.

a, Timeline of infection and number of tetramer+CD8+ T cells isolated from each sample. b, FACS plots and precursor frequency of tetramer+CD8+ T cells before, during and after IBV infection. c, Principal component analysis of tetramer+CD8+ T cells sequenced. Time points are distinguished by color and specificity by shape. d, Heatmap illustrating expression of differentially expressed genes identified across all the time points compared to the baseline as reference using MAST. Cells grouped by epitope and time point. e, Heatmap representing gene set enrichment of upregulated (pink) and downregulated (green) genes of tetramer+CD8+ T cells sorted at day 14 compared to baseline. f, Heatmap representing gene set enrichment of upregulated (pink) and downregulated (green) genes of tetramer+CD8+ T cells sorted at day 14 compared to 1.5-year time point.
A total of 209 tetramer+CD8+ T cells were analyzed using scRNA-seq, with ~1,201 expressed genes identified per cell. Principal component analysis revealed clear segregation of A2-PB1413+CD8+ T cells by time point but no segregation between the two antigenic specificities on day 14 (Fig. 5c). Notably, differential expression analysis identified distinct gene expression signatures across time points (Fig. 5d). Gene-set enrichment analysis showed that signatures of T cell activation and differentiation, cell division, immune cell migration and chemotaxis were enriched in day 14 cells as compared to those from baseline or 1.5 years (Fig. 5e,f).
We next analyzed the expression of specific genes associated with T cell differentiation, activation, cytotoxicity and effector function (Supplementary Fig. 6c,d). Importantly, effector CD8+ T cells across both IBV specificities isolated from day-14 upregulated genes associated with activation, cytotoxic molecules, cytotoxic receptors and effector cytokines. The expression profiles for selected genes associated with differentiation and activation were confirmed by flow cytometry (Supplementary Fig. 6c,d).
Although single-cell RNA-seq data were obtained from one subject naturally infected with IBV, this experiment provided a rare opportunity to examine baseline PBMC samples from a HLA-A*0201-expressing subject before natural IBV infection, at the acute (day 14), short-term memory (3 months) and long-term memory (1.5 years) time points after IBV. Our results provide evidence of transcriptome changes associated with differentiation and activation of A2-PB1413+CD8+ and A2-BHA543+CD8+ T cells during IBV infection. To the best of our knowledge, these are the first data on transcriptome changes within tetramer-specific CD8+ T cells at the single-cell level from the baseline to long-term memory CD8+ T cells in humans. Thus, through the flow cytometric analysis of IBV-infected subjects and longitudinal scRNA-seq analysis of a naturally infected individual, we demonstrate that A2-PB1413+CD8+ and A2-BHA543+CD8+ T cells are recruited to the immune response during IBV infection.
BHA543 + and BNS1266 +CD8+ T cells in IBV-infected mice
Having shown recruitment of activated CD8+ T cells against universal (A2-PB1413) and novel IBV-specific (A2-BHA543) epitopes during influenza in humans, we subsequently investigated the protective efficacy of these cells, especially as the role of CD8+ T cells in IBV infection remains unclear. To achieve this, we used HLA-A2.1-expressing transgenic (HHD-A2) mice previously used for IAV infection38, and established a HHD-A2 mouse model of IBV and ICV infection. HHD-A2 mice express a chimeric MHC-I monochain comprising human β2-microglobulin covalently linked to the HLA-A*02:01 α1 and α2 domains, and murine α3 and transmembrane domains39, and thus can respond to many human HLA-A*02:01-restricted epitopes, including IAV-derived A2-M158 (ref. 38) and cancer-derived A2-WT1A neoantigen40. These mice are not confounded by infection history or coexpression of other MHC-I molecules and provide a tool for understanding influenza-specific CD8+ T cells in vivo and determining their protective role in influenza.
To verify the immunogenicity of novel IBV-derived peptides, we infected HHD-A2 mice intranasally with B/Malaysia. On day 10 after infection, we stimulated splenocytes with the 67 novel IBV peptides individually and measured IFN-γ and ΤΝF production. As in humans (Fig. 2h), CD8+ T cell responses were targeted towards A2-BHA543–551 (mean 5% of CD8+ T cells) and A2-BNS1266–274 (mean 1.8%), with smaller responses observed for A2-BHA538–551 and A2-BNS1264–274 (mean <0.5%), which overlap with A2-BHA543–551 and A2-BNS1266–274, respectively (Fig. 6a,b). We also assayed six pools of 10–12 peptides, as for humans (Supplementary Table 2), and assessed CD8+ T cells to those at the site of infection (bronchoalveolar lavage, BAL). CD8+ T cell responses were targeted to pools 2 and 3, which contained the BHA543–551, BNS1266–274 and BNS1264–274 peptides, as confirmed separately (Fig. 6c,d).

a, Representative FACS plots for immunogenic peptides. b, Frequency of IFN-γ+TNF+CD8+ T cells of total CD8+ T cells in the spleen of IBV-infected mice toward each peptide. Mean and s.e.m. are shown (n = 8 from at least two independent experiments). c,d, CD8+ T cell responses in the BAL. c, Cytokine responses to each peptide pool. Data from two independent experiments in which the BAL of multiple (n = 3 or 5 per experiment) mice were pooled. d, Cytokine responses to individual immunogenic peptides in the BAL (n = 4 from one experiment). Mean and s.e.m. are shown.
To compare primary CD8+ T cells directed at BHA543–551 and BNS1266–274 epitopes with secondary responses, we primed HHD-A2 mice intranasally with B/Malaysia, and then intranasally infected them with the heterologous strain B/Phuket 6 weeks later (Supplementary Fig. 7b,c). Assessment of CD8+ T cell responses against the main A2-BHA543–551 and A2-BNS1266–274 epitopes on day 8 after challenge showed that the number of secondary IFN-γ+TNF+CD8+ T cells in the spleen was ~27-fold higher than that after primary infection (Supplementary Fig. 7a). Additionally, CD8+ T cells for both specificities showed increased polyfunctionality (IFN-γ+TNF+IL-2+) after challenge (0.14 and 2.14%, respectively, for BHA543) (Supplementary Fig. 7d). Thus, using our model of IBV in HHD-A2 mice, we verified the novel (identified by immunopeptidomics) immunodominant IBV-specific A2-BHA543–551 and A2-BNS1266–274 epitopes in both primary and secondary IBV infections.
Lack of A2-PB1413 +CD8+ T cells in HHD-A2 mice
As universal A2-PB1413+CD8+ T cells can be detected in both IAV- and IBV-infected subjects, we assessed A2-PB1413+CD8+ T cells after IAV (A/X31), IBV (B/Malaysia) or ICV (C/Perth) infection of HHD-A2 mice. To our surprise, CD8+ T cells specific for the A2-PB1413 epitope were not detected after (1) primary IAV, IBV or ICV infection (Supplementary Fig. 8a), (2) secondary infection with a heterologous virus (for example, A/X31→A/PR8) or a heterotypic virus (for example, A/X31→B/Mal) in all four possible combinations (A→A, A→B, B→B and B→A) (Supplementary Fig. 8b) or (3) tertiary (A→B→A, B→A→B) (Supplementary Fig. 8c) influenza infections (Supplementary Results). Additionally, A2-PB1413+CD8+ T cells in HHD-A2 mice were not detected after lipopeptide (Supplementary Fig. 8d) or peptide (Supplementary Fig. 8f) vaccination or with tetramer enrichment in naive mice. Thus, the above experiments (Supplementary Fig. 8) provide strong evidence for a lack of naive A2-PB1413-specific precursors in HHD-A2 mice, probably owing to a T cell antigen receptor (TCR) repertoire hole in HHD-A2 mice toward the A2-PB1413 epitope. Hence, the protective role of universal A2-PB1413+CD8+ T cells toward IAV, IBV and ICV infection could not be assessed in HHD-A2 mice (Supplementary Note).
Protective capacity of BHA543 + and BNS1266 +CD8+ T cells in HHD-A2 mice
Previous studies using CD8+ T cell depletion in mice lacking antibodies have demonstrated a role for CD8+ T cells during IBV infection41. To determine the protective capacity of novel IBV-derived CD8+ T cell epitopes in HHD-A2 mice, we vaccinated mice with the BHA543 and BNS1266 peptides using a prime-boost approach, then infected mice intranasally with 5 × 103 plaque-forming units (p.f.u.) B/Malaysia (Fig. 7a). On day 6 after boosting and before vaccination, numbers of innate cells (neutrophils, macrophages and γδ T cells) were comparable between mock (adjuvant alone) and peptide-adjuvant-vaccinated groups in blood (data not shown). Thus, any nonspecific inflammatory or innate effects of vaccination are controlled for in mock animals.

a, Detailed experimental plan of vaccination. b–d, Tetramer-specific CD8+ T cells responses on days 6 and 7 after infection in BAL and spleen. b, Representative FACS plots. c, Number of total (A2-BHA543 and A2-BNS1266) tetramer+ CD8 T cells in the spleen on day 7 after IBV infection. d, Number of individual A2-BHA543+ and A2-BNS1266+ tetramer+ CD8+ T cells in the spleen and BAL on days 6 (D6) and 7 (D7) after IBV infection. e, Viral titers in lungs and nose of peptide-vaccinated and mock-vaccinated mice after IBV infection. Days 5 (n = 5) and 6 (n = 5) were assessed in an independent experiment to day 7 (n = 4–5). f,g, Cytokine responses in the BAL on day 7 after IBV challenge (n = 4–5). Throughout the figure, means and s.e.m. are shown (n = 5 mice per group, except from mock group day 7 where n = 4). Data from days 6 and 7 are from two independent experiments. For c,d, statistical significance was determined using a two-way analysis of variance (Sidak’s multiple comparisons). c, **P = 0.002; d, **P = 0.0014, *P = 0.016. For e–g, statistical significance was determined using an unpaired two-tailed t-test. e, Lung, *P = 0.016, **P = 0.005; nose, **P = 0.0076; g,*P = 0.0038, #P = 0.024, **P = 0.0075.
Vaccination with peptides led to significantly higher numbers of total A2-BHA543- and A2-BNS1266-tetramer+CD8+ T cells in the spleen on days 6 and 7 after IBV infection when compared to mock-vaccinated (adjuvant alone) mice (~5.6-fold) (Fig. 7b,c). A2-BHA543+CD8+ and A2-BNS1266+CD8+ T cell numbers were comparable (P > 0.05) in the BAL (about two-fold increase in immunized mice). After immunization, however, there was an increase in recruitment of A2-BHA543+CD8+ T cells to the site of infection between days 6 and 7 (Fig. 7d). Importantly, peptide-vaccinated mice exhibited significant protection against IBV, as shown by a significant ~65% reduction in viral titers in the lung and nose on day 6 and 100% clearance in the lung on day 7 after IBV infection when compared to the mock-immunized group (Fig. 7e). Additionally, there was a significant decrease in inflammatory cytokines (MIP-1β, IL-6, IL-1β and IFN-γ) in day 7 BAL of peptide-vaccinated mice in comparison to mock-immunized animals (Fig. 7f,g). Thus, CD8+ T cells directed at our novel HLA-A2.1-restricted IBV-specific epitopes are protective, as they can markedly accelerate viral clearance and reduce the cytokine storm at the site of infection.
Discussion
Cytotoxic CD8+ T cells have a crucial role in protection from severe influenza disease in humans and animal models of influenza virus infection1,2. CD8+ T cells limit viral replication and promote clearance of infected cells whose recognition depends on presentation of viral peptides by MHC-I molecules. High conservation of these peptides allows cross-recognition of distinct IAV strains, including pandemic and avian IAV viruses1,2. Our study examines two levels of cross-reactivity by influenza-specific CD8+ T cells: (1) heterotypic cross-reactivity across IAV and IBV, and in some instances ICV, by CD8+ T cells recognizing peptides derived from conserved influenza regions, and (2) IBV-wide cross-reactivity by CD8+ T cells recognizing conserved IBV regions.
Broadly neutralizing antibodies to the IAV and/or IBV hemagglutinin stem are the focus of recent research. However, so far, such broadly cross-reactive antibodies are rare and immuno-subdominant compared to strain-specific antibodies to the variable hemagglutinin head7. Conversely, broadly cross-reactive CD8+ T cells are abundant at the population level, can be recruited in ~50% and ~80% of HLA-A*02:01-expressing individuals infected with IAV or IBV, respectively, and can account for substantial immune responses towards influenza. Thus, combining broadly neutralizing antibodies with broadly cross-reactive and abundant CD8+ T cells is important for optimal universal protection against distinct influenza strains. Cross-reactivity across IAV and IBV is unprecedented for CD8+ T cells and atypical for influenza-specific CD4+ T cells and antibodies. Only one rare antibody (CR9114) cross-recognizing conserved IAV and IBV hemagglutinin stem regions has been reported8 and its contribution to human infection remains unknown. Similarly, a highly conserved CD4+ T cell epitope containing a peptide from the hemagglutinin fusion peptide has been identified although it has been poorly characterized42. Universal memory A2-PB1413 CD8+ T cells, however, are prominent in human blood and lung tissues, and emerge as activated effectors during human IAV and IBV. Additionally, such CD8+ T cells were found in the majority (80%) of donors we tested, and thus are abundant across HLA-A*02:01+ donors. Furthermore, our reported heterotypic cross-reactivity, restricted by HLA-A*02:01, HLA-A*01:01 and HLA-B*37:01, covers ~54% of the world population.
The IBV-wide cross-reactivity resembles IAV-wide cross-reactivity provided by well-characterized CD8+ T cells, exemplified by A2-M158 (ref. 38). Although the ability of CD8+ T cells to cross-react across IBV lineages has been reported11, the antigenic specificity underpinning such cross-reactivity has been unknown. We identified CD8+ T cell targets from BHA and BNS1 proteins and show that these responses are protective in mice. The observation that IBV-wide cross-reactivity can occur towards peptides derived from BHA is intriguing, as it contests the belief that CD8+ T cell cross-reactivity is conferred by peptides from internal influenza proteins. Given the high prevalence of HLA-A*02:01 and the clinical significance of IBV, our work implies that CD8+ T cell–targeting vaccines should be formulated with broader antigenic specificity not limited to NP and M1.
Here, we provide direct evidence for the role of CD8+ T cells in protection against IBVs, as demonstrated by immunization with our newly identified IBV peptides. After IBV infection, peptide-immunized mice displayed significant reduction in viral titers and inflammation when compared to mock-immunized animals. To our surprise, A2-PB1413+CD8+ T cells were undetectable in HHD-A2 mice, irrespective of the infection or immunization protocol. This indicates that HHD-A2 mice lack naive A2-PB1413+CD8+ TCR precursors, probably resulting from a TCR repertoire hole, consistent with previous studies in HHD-A2-DRB1 mice43. Although HHD-A2 mice express a chimeric MHC-I monochain comprising the human β2-microglobulin covalently linked to HLA-A*02:01 α1 and α2 domains and the murine α3 and transmembrane domains, TCRs remain murine. Thus, although HHD-A2.1-expressing mice respond to several human HLA-A*02:01-restricted epitopes38,40,44 TCRs for others might be lacking. Although these mice are useful for screening peptides, such results should be validated in human samples. HHD mice are, however, important in assessing the protective capacity of HLA-A2-restricted CD8+ T cells. By vaccinating HHD-A2 mice and challenging them with IBV, we provide evidence for a protective role of CD8+ T cells in IBV infection after peptide vaccination, consistent with CD8+ T cell depletion in mice lacking B cells41. In conjunction with the activation of these CD8+ T cells in subjects and the presence of TRM cells in the human lung, our studies suggest that vaccinating against these epitopes could provide protection in humans.
The antigenic origin of broadly cross-reactive epitopes is of interest. PB1 is the most conserved protein across IAV and IBV, with ~60% amino acid identity, in contrast with 30% for other proteins4,24. The PB1413 peptide is derived from the most conserved protein region, motif B (residues 406–422 of IAV-PB1 protein), a core motif present in viral RNA-dependent polymerases. Genome-wide mutational analysis has demonstrated that IAV cannot tolerate substitutions in these motifs45. Notably, the IBV-wide cross-reactivity is conferred by the BHA543 peptide, which is derived from the BHA stalk, which shows considerably higher conservation than the hemagglutinin head46. Mutagenesis screens have revealed limited tolerance to 15-nucleotide insertions in BHA, particularly in the stalk domain46. Thus, universally cross-reactive CD8+ T cells target epitopes with little sequence flexibility, making them ideal targets for universal and IBV-wide influenza vaccines.
The ability of CD8+ T cells to confer heterotypic cross-reactivity across IAV and IBV and the knowledge of cross-reactive epitopes across IAV and IBV types and within IBV strains have implications for the design of universal influenza vaccines that do not require annual reformulation. Pre-emptive influenza vaccines eliciting broadly cross-reactive and long-lasting CD8+ T cells would reduce annual rates of IAV- and IBV-induced morbidity and mortality globally. Additionally, influenza vaccines eliciting immunity across IAV, IBV and ICV could protect children from severe ICV disease5. Furthermore, T cell–targeted vaccines would augment numbers of universal IAV-, IBV- and ICV-specific CD8+ T cells and IAV- and IBV-specific CD8+ T cells in individuals with previous viral exposures and thus confer stronger protective immunity after infection14. It is important to consider universal CD8+ T cells alongside universal antibodies for the design of universally cross-reactive influenza vaccines, especially as current inactivated influenza vaccines do not elicit influenza-specific CD8+ T cell responses19.
Methods
Cell lines, viruses and reagents
C1R cells (parental or stable transfectants expressing specific HLA alleles; a gift from W. Chen, La Trobe University) were maintained in RF10 medium (RPMI-1640 with 10% heat-inactivated FCS, 1 mM MEM sodium pyruvate, 2 mM l-glutamine, 100 mM MEM nonessential amino acids, 5 mM HEPES buffer solution, 55 mM 2-mercaptoethanol, 100 U ml–1 penicillin and 100 mg ml–1 streptomycin; purchased from Gibco (Thermo Fisher Scientific)). Influenza A (A/Switzerland/9715293/2013, A/X31 and A/PR8) and B (B/Malaysia/2506/04 and B/Phuket/3073/2013) viruses were grown in the allantoic cavity of day 10–embryonated chicken eggs for 3 d at 35 oC and viral titers were determined by plaque assay on MDCK cells (American Type Culture Collection). Influenza B viruses were provided by Seqirus. Influenza C viral isolates (C/Perth/08/2012) were provided by the World Health Organization Collaborating Centre for Reference and Research on Influenza (Melbourne, Australia). The BNP-FITC (1:200, H89B, ThermoFisher) antibody was used to assess infection levels with IBV. Synthetic peptides were purchased from GenScript and reconstituted in 10–100% DMSO Hank’s Balanced Salt Solution. Tetramers were generated by first producing soluble HLA α-heavy chain-BirA and β2-microglobulin (β2-m) before refolding with peptide to generate monomers. Monomers were conjugated to fluorescent-labeled streptavidins (SA) such as PE-SA, APC-SA or BV421-SA (BD Biosciences), at an 8:1 monomer to SA molar ratio to form pMHC-I tetramers. Cell lines were confirmed to be mycoplasma free using the MycoAlert Mycoplasma Detection Kit (Lonza).
Human blood and tissue samples
Human experimental work was conducted according to the Declaration of Helsinki Principles and according to the Australian National Health and Medical Research Council Code of Practice. Signed informed consents were obtained from all blood and tissue donors before the study. Spleen, lymph node and lung tissues were obtained from deceased organ donors after written informed consent from the next of kin. Spleen and lymph node samples were obtained via DonateLife Victoria. Lung samples were obtained via the Alfred Hospital’s Lung Tissue Biobank from individuals who did not die of immunological conditions or influenza infection. Tonsils were obtained from healthy individuals undergoing tonsillectomy (Mater Hospital, North Sydney, Australia). PBMCs were isolated from buffy packs obtained from the Australian Red Cross Blood Service (ARCBS, West Melbourne, Australia). Human blood and tissue samples were processed as described19. The study was approved by the University of Melbourne Human Ethics Committee (1443389.4 and 1545216.1), the ARCBS Ethics Committee (2015#8), the Alfred Hospital Ethics Committee (280/14), Monash Health Human Research Ethics Committee (HREC/15/MonH/64, RMH local reference number 2016/196), St. Jude Children’s Research Hospital (XPD12–089 IIBANK), the University of Tennessee Health Science Center/Le Bonheur Children’s Hospital, and the Human Research Ethics Committee, Research and Ethics Governance Office of the Royal Prince Alfred Hospital, Sydney Local Health District (X13–0372).
Healthy donors and clinical cohorts
The characteristics of the healthy PBMC donors and organ donors are described in Supplementary Tables 3–7. Influenza-infected and influenza-negative adult subjects were recruited from the Alfred Hospital and Royal Melbourne (Melbourne, Australia). Informed consent was obtained before sampling. Influenza infection was confirmed by PCR. Pediatric and adult subjects were recruited at St. Jude Children’s Research Hospital (Memphis, TN, USA), as part of the FLU09 cohort, which has been described47,48. Written, informed consent was acquired from participants’ parents or guardians and written assent from age-appropriate subjects was acquired at the time of enrollment. The characteristics of the influenza-infected and influenza-negative donors are described in Supplementary Tables 8 and 9, respectively.
HLA-A2.1 HHD mouse studies
HHD-A2 mice express a chimeric MHC-I monochain comprising human β2-microglobulin covalently linked to the HLA-A*02:01 α1 and α2 domains and the murine α3 and transmembrane domains39. HHD-A2 mice were developed by F. Lemonnier and the parental pairs were provided by the Pasteur Institute. Mice aged 6–12 weeks were used. All animal work was conducted in accordance with guidelines set by the University of Melbourne Animal Ethics Committee (ethics approval numbers 1312880.4 and 1714108.5). HHD-A2 mice were infected intranasally with 30 μl of influenza virus in PBS under isoflurane anesthesia. For IAV, mice were infected with 100 p.f.u. of X31 or PR8, IBV with 100 p.f.u. of B/Malaysia or B/Phuket, and ICV with 50 μl of C/Perth/8/2012 with an hemagglutinin titer of 64. Primary, secondary and ternary infections were performed 6–8 weeks apart. For peptide vaccination, 30 nmol of each peptide was mixed and emulsified in complete (for priming) or incomplete (for boosting) Freund’s adjuvant and delivered subcutaneously at the base of the tail (50 μl on either side). Mice were boosted 2 weeks after priming and challenged with 5 × 103 p.f.u. of B/Malaysia intranasally 7 d after boosting. Viral titers were determined from lung and nose homogenates using plaque assays as described49. Cytokines were measured on lung homogenates and BALF using a cytometric bead array kit (BD Biosciences) as described49. For lipopeptide vaccination, 20 nmol of Pam2Cys-lipoptide with the PB1413 or M158 peptides was delivered intranasally in 50 μl of saline under isoflurane anesthesia.
Detection of naive epitope–specific CD8+ T cells was performed as described50. Briefly, the spleen and major lymph nodes (auxiliary, brachial, cervical, inguinal, mesenteric and mediastinal) were pooled and processed into single-cell suspensions for each mouse. Cells were stained with tetramers conjugated to PE or APC for 1 h at room temperature. Tetramer+ cells were enriched using anti-PE or anti-APC MicroBeads (Miltenyi Biotec) and an LS column (Miltenyi Biotec) according to the manufacturer’s instructions. Cells were then stained with Fixable Live/Dead AquaBlue viability dye (Life Technologies) and surface markers. Tetramer+CD8+ T cells were identified as CD8α+CD3e+CD11c−CD11b−F4/80−B220−I-Ab–NK1.1−CD4−). The whole sample was acquired by FACS.
Viral peptide conservation analysis
Unique amino acid sequences of the eight segments of influenza A, B and C viruses (n = 56,353, 10,235 and 456, respectively) were downloaded from the National Center for Biotechnology Information database51, using full-length sequences from any host, any location and any subtype. Sequences were screened for the presence of A- and B-epitopes using the Smith-Waterman search algorithm, which detects the best position of a query within a longer string while allowing for some single-amino acid differences. To assess conservation of a given viral peptide, we analyzed two metrics: whether or not a “match” was found for that epitope in the search targets, where we classified a match as a search result ≥70% similar to the query epitope, and the overall average percentage similarity of the matches returned. To analyze temporal variation in epitope conservation of two select epitopes (BHA543–551 and BNS1266–274), all influenza BHA and NS1 data from Global Initiative on Sharing All Influenza Data along with sample collection dates were downloaded. Minimum lengths of 550 amino acids for the HA and 250 amino acids for the NS1 were selected, providing 16,500 records for HA and 14,845 sequences for NS1. Similar to prior analysis, the Smith-Waterman algorithm at a threshold of ≥70% similarity was used to screen for positive hits. The code used for this analysis is available upon request.
Expansion of antigen-specific memory CD8+ T cells from human PBMCs
Cryopreserved PBMCs (~1–5 × 106) from healthy adults were used to expand antigen-specific T cells as described52. Briefly, to expand peptide-specific CD8+ T cells, one-third of PBMCs were pulsed with 1–10 μM of peptide in serum-free RPMI for 1 h at 37 °C, washed twice and then mixed with the remaining autologous PBMCs. To expand virus-specific CD8+ T cells, one-tenth of PBMCs were infected with IAV or IBV (B/Malaysia) in serum-free RPMI for 1 h at 37 °C, washed twice and then mixed with the remaining autologous PBMCs. Cells were then incubated for 9–12 d in RF10 medium with 10 U ml–1 of recombinant human IL-2 (Roche Diagnostics) added on day 4 and half-media changes every 2 d onward.
T cell re-stimulation and intracellular cytokine staining
To measure ex vivo ICS responses, PBMCs were thawed and rested overnight in RF10 medium with 10 U ml–1 of recombinant human IL-2. Between 0.8 × 106 and 8 × 106 cells were stimulated with 1 μM peptide for 6 h in the presence of brefeldin A (BD GolgiPlug) and monensin (BD GolgiStop). For in vitro T cell lines, day 9–12 PBMC cultures were restimulated with 1 μM peptide for 5 h in the presence of brefeldin A (BD GolgiPlug) and monensin (BD GolgiStop). In some experiments, PBMCs were restimulated with peptide-pulsed HLA-expressing C1R cells. After stimulation, cells were surface and intracellularly stained as described below with panel 1 (Supplementary Table 10).
Identification of epitope-specific CD8+ T cells by tetramer staining
PBMCs were incubated with pMHC-I tetramers (A2-M158–66, A2-PA46–54, A2-PB1413–421, A2-BHA543–551 and A2-BNS1266–274) for 1 h in MACS buffer (PBS with 0.5% BSA and 2 mM EDTA) at room temperature. After two washes, surface antibody staining was performed in 50 µl MACS buffer for 20 min at 4 °C.
Immunopeptidome analysis
Cell pellets of 8 × 108 to 10 × 108 cells were lysed using a combination of mechanical and detergent-based lysis. The lysates were cleared by ultracentrifugation and MHC complexes were isolated by immunoaffinity purification using solid-phase-bound monoclonal antibodies for immunoaffinity purification as described53. Antibodies BB7.2 (anti-HLA-A2) and w632 (anti-pan class I) were used sequentially for purification of transfected HLA-A*02:01 and remaining endogenous MHC class I (marginal HLA-B*35:03 and HLA-C*04:01) of the C1R cells. LB3.1 (anti-HLA-DR), SPV-L3 (anti-HLA-DQ) and B721 (anti-HLA-DP) were subsequently used to isolate MHC class II complexes. Peptides were eluted from the MHC with 10% acetic acid and fractionated on a 4.6-mm internal diameter × 100-mm monolithic reverse-phase C18 high-performance liquid chromatography (HPLC) column (Chromolith SpeedROD; Merck Millipore) using an ÄKTAmicro HPLC (GE Healthcare) system, running a mobile phase consisting of buffer A (0.1% trifluoroacetic acid; Thermo Fisher Scientific) and buffer B (80% acetonitrile, 0.1% trifluoroacetic acid; Thermo Fisher Scientific). MHC-peptide mixtures were loaded onto the column at a flow rate of 1 ml min–1 (2% buffer B, 98% buffer A) and separated at 2 ml min–1 based on a buffer B gradient of increasing buffer B content from 2 to 15% over 0.25 min, 15–30% over 4 min, 30–40% over 8 min and 40–45% over 10 min before the flow rate was reduced to 1 ml min–1 and buffer B rapidly increased 45–99% over 2 min. We collected 500-µl fractions throughout, and the peptide-containing fractions were combined into nine pools, vacuum-concentrated and reconstituted in 12 µl 0.1% formic acid (Thermo Fisher Scientific).
Reconstituted fraction pools were analyzed by LC–MS/MS using an information-dependent acquisition strategy on a Q-Exactive Plus Hybrid Quadrupole Orbitrap (Thermo Fisher Scientific) coupled to a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). 6 µl of concentrated material was loaded onto a Dionex Acclaim PepMap100 200-mm C18 Nano-Trap Column with 100-μm internal diameter (5-μm particle size, 300-Å pore size) in buffer A (2% acetonitrile, 0.1% formic acid) at a flow rate of 15 µl min–1. Peptides were then separated by switching a Dionex Acclaim RSLC PepMap RSLC C18 column (50-cm length, 75-μm internal diameter, 2-μm particle size, 100-Å pore size) in line and eluting the peptides (250 nl min–1) over an increasing gradient of buffer B (80% acetonitrile, 0.1% formic acid) of 2.5–7.5% over 1 min, 7.5–35% over 40 min, 35–99% over 5 min, 99% over 6 min and returning to 2.5% buffer B over 1 min, before re-equilibration at 2% for 20 min. Data were collected in positive mode: MS1 resolution, 70,000; scan range, 375–1,800 m/z; MS2 resolution, 17,500; scan range, 200–2,000 m/z. The top 12 ions of +2 to +5 charge per cycle were chosen for MS/MS with a dynamic exclusion of 15 s. Raw files were converted to mgf format and spectra were searched against a proteome database consisting of the human proteome (UniProt/Swiss-Prot v2016_04), the B/Malaysia proteome and a six-reading frame translation of the B/Malaysia genome using ProteinPilot software (version 5.0, SCIEX), considering biological modifications and using a decoy database for false discovery rate analysis. Length distribution and motif analyses were based on human peptides assigned at confidences greater than that required for a 5% false discovery rate, precursor delta mass ≤ 0.05. Peptides assigned to the influenza proteome at any confidence were also considered, precursor delta mass ≤ 0.05. Probable HLA-A*02:01 binders were determined based on appearance across the experiments and antibodies (BB7.2 versus w632 and class II). Of the 73 ligands, 67 were chosen for synthesis in native form and used in subsequent analyses. Selected peptides met any of the following criteria: (1) identification in both experiments, (2) confidence of sequence assignment >50, (3) predicted half-maximum inhibitory concentration (IC50) of binding to HLA-A*02:01 <2,500 nM. The 12-mers to 14-mers were synthesized regardless of meeting these criteria. The HLA-A*02:01 binding predictions were made on 18.07.2018 using the Immune Epitope Database Analysis Resource ANN, also known as NetMHC (ver. 4.0), tool54,55,56. Peptide binding motifs were visualized using Icelogo software (static reference method against the SWISS-PROT human proteome)57.
Screening of novel peptide ligands
For in vivo screening, HHD mice were infected with 100 p.f.u. of B/Malaysia intranasally. On day 10, spleen and BAL were harvested and processed into single-cell suspensions. Lymphocytes were stimulated with 1 μM peptide in RF10 with 10 U ml–1 of recombinant human IL-2 for 5 h in the presence of brefeldin A (BD GolgiPlug). Cells were stained with panel 3 (Supplementary Table 10) and analyzed as described above. For in vitro screening, cryopreserved PBMCs from HLA-A*02:01+ healthy adults were expanded with peptide pools (final concentration of 10 μM). Responses against cognate pools were assessed on day 9–10. Any positive pools were further dissected by restimulating remaining cultures with individual peptides on day 11–12. Immunogenic peptides were validated by in vitro expansion with single peptide and ICS assay with the C1R-A2 cells pulsed with the cognate peptide.
Tetramer-associated magnetic enrichment of antigen-specific CD8+ T cells
TAME was performed as described29. Briefly, cryopreserved PBMCs (1 × 106 to 50 × 106) were thawed, counted and incubated with anti-human FcR block (Miltenyi Biotec) before being stained with pMHC-I tetramers conjugated to PE-SA or APC-SA for 1 h at room temperature. Cells were washed, incubated with anti-PE or anti-APC MicroBeads (Miltenyi Biotec) and passed through a LS column (Miltenyi Biotec) according to the manufacturer’s instructions. After magnetic enrichment, unenriched, flow-through and enriched samples were stained for surface and intracellular markers as described above. A limit of detection for TAME analysis was set at 10–7, as determined by Alanio et al27. Panels 4a and 4b were used (Supplementary Table 10). Lung samples were not enriched and were stained with panel 5.
Single-cell RNA-seq
Single tetramer+ cells were sorted into 96-well plates containing 0.5 μl dNTP mix (10 mM), 0.5 μl oligo-dT primer (5 μM), and 1 μl lysis buffer (prepared by adding 1 μl RNase inhibitor to 19 μl Triton X-100 solution, 0.2% (v/v)). Libraries were prepared using the Smart-seq2 protocol as described18,58,59 with the following modifications. cDNA synthesis and preamplification reaction volumes were halved to 5 and 12.5 μl, respectively. The final concentration of the ERCC RNA Spike-In mix in the reverse-transcription step was 1:400 million. Sequencing libraries were prepared using Nextera XT DNA Library Prep Kit and sequenced using on a NextSeq 500 platform with 150-base pair high-output paired-end chemistry.
Bioinformatics analysis
The scRNA-seq data were preprocessed for quality control. Reads had an average length of 150 base pairs and each cell had an average sequencing depth of 1 million paired-end reads, enough to robustly assess gene expression and reconstruct TCR sequences from scRNA-seq. The quality of scRNA-seq reads was assessed using FastQC and reads were aligned to Ensembl GRCh37 reference genome using TopHat2 with default parameters. Only cells that passed initial filtering were retained for downstream analysis. One cell with a low number of genes expressed (<400 genes with at least 10 fragments per kilobases per million (FPKM)) was excluded from the analysis. The Cufflinks suit (v2.2.1) was used to calculate gene expression in FPKM; with CuffQuant used to calculate FPKM and CuffNorm to normalize the FPKM values based on total mRNA content. Differential expression analysis and gene set enrichment analysis were performed as indicated in MAST tutorial. Heatmaps were generated with the R package pheatmap.
Statistical analysis
Statistical significance of nonparametric data sets was determined by the Mann-Whitney test for unpaired comparisons and a two-tailed Wilcoxon matched-pairs signed-rank test for paired comparisons. An unpaired t-test was used for parametric data sets. A two-way analysis of variance (Sidak’s multiple comparisons) was used to compare tetramer-specific responses in mice across groups and time points.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. scRNA-seq data that support the findings of this study have been deposited in Arrayexpress with accession code E-MTAB-7606.
References
Krammer, F. et al. Influenza. Nat. Rev. Dis. Primers 4, 3 (2018).
Nussing, S. et al. Innate and adaptive T cells in influenza disease. Front. Med. 12, 34–47 (2018).
Paules, C. & Subbarao, K. Influenza. Lancet 390, 697–708 (2017).
Koutsakos, M., Nguyen, T. H., Barclay, W. S. & Kedzierska, K. Knowns and unknowns of influenza B viruses. Future Microbiol. 11, 119–135 (2016).
Matsuzaki, Y. et al. Clinical features of influenza C virus infection in children. J. Infect. Dis. 193, 1229–1235 (2006).
Chen, Y. Q. et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell 173, 417–429 e410 (2018).
Corti, D. et al. Tackling influenza with broadly neutralizing antibodies. Curr. Opin. Virol. 24, 60–69 (2017).
Dreyfus, C. et al. Highly conserved protective epitopes on influenza B viruses. Science 337, 1343–1348 (2012).
Hayward, A. C. et al. Natural T cell-mediated protection against seasonal and pandemic influenza. Results of the Flu Watch Cohort Study. Am. J. Respir. Crit. Care. Med. 191, 1422–1431 (2015).
McMichael, A. J., Gotch, F. M., Noble, G. R. & Beare, P. A. Cytotoxic T-cell immunity to influenza. New Engl. J. Med. 309, 13–17 (1983).
van de Sandt, C. E. et al. Influenza B virus-specific CD8+T-lymphocytes strongly cross-react with viruses of the opposing influenza B lineage. J. Gen. Virol. 96, 2061–2073 (2015).
Gras, S. et al. Cross-reactive CD8+T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc. Natl Acad. Sci. USA 107, 12599–12604 (2010).
Greenbaum, J. A. et al. Pre-existing immunity against swine-origin H1N1 influenza viruses in the general human population. Proc. Natl Acad. Sci. USA 106, 20365–20370 (2009).
Quinones-Parra, S. M. et al. A role of influenza virus exposure history in determining pandemic susceptibility and CD8+ T cell responses. J. Virol. 90, 6936–6947 (2016).
Sridhar, S. et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 19, 1305–1312 (2013).
Quinones-Parra, S. et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc. Natl Acad. Sci. USA 111, 1049–1054 (2014).
Wang, Z. et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nat. Commun. 6, 6833 (2015).
Wang, Z. et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat. Commun. 9, 824 (2018).
Koutsakos, M. et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci. Transl. Med. 10, eaan8405 (2018).
Ramarathinam, S. H. et al. Identification of native and posttranslationally modified HLA-B*57:01-restricted HIV envelope derived epitopes using immunoproteomics. Proteomics 18, e1700253 (2018).
Williamson, N. A. & Purcell, A. W. Use of proteomics to define targets of T-cell immunity. Expert. Rev. Proteomics 2, 367–380 (2005).
Alexander, J. et al. Identification of broad binding class I HLA supertype epitopes to provide universal coverage of influenza A virus. Hum. Immunol. 71, 468–474 (2010).
Rosendahl Huber, S. K. et al. Chemical modification of influenza CD8+ T-cell epitopes enhances their immunogenicity regardless of immunodominance. PLoS ONE 11, e0156462 (2016).
Terajima, M., Babon, J. A., Co, M. D. & Ennis, F. A. Cross-reactive human B cell and T cell epitopes between influenza A and B viruses. Virol. J. 10, 244 (2013).
Zemmour, J., Little, A. M., Schendel, D. J. & Parham, P. The HLA-A,B “negative” mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J. Immunol. 148, 1941–1948 (1992).
Falk, K., Rotzschke, O., Stevanovic, S., Jung, G. & Rammensee, H. G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296 (1991).
Alanio, C., Lemaitre, F., Law, H. K., Hasan, M. & Albert, M. L. Enumeration of human antigen-specific naive CD8+T cells reveals conserved precursor frequencies. Blood 115, 3718–3725 (2010).
Moon, J. J. et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213 (2007).
Nguyen, T. H. O. et al. Perturbed CD8(+) T cell immunity across universal influenza epitopes in the elderly. J. Leukoc. Biol. 103, 321–339 (2018).
Chandele, A. et al. Characterization of human CD8 T Cell responses in Dengue virus-infected patients from India. J. Virol. 90, 11259–11278 (2016).
Jozwik, A. et al. RSV-specific airway resident memory CD8+T cells and differential disease severity after experimental human infection. Nat. Commun. 6, 10224 (2015).
Miller, J. D. et al. Human effector and memory CD8+T cell responses to smallpox and yellow fever vaccines. Immunity 28, 710–722 (2008).
Hillaire, M. L. et al. Characterization of the human CD8(+) T cell response following infection with 2009 pandemic influenza H1N1 virus. J. Virol. 85, 12057–12061 (2011).
de Bree, G. J. et al. Selective accumulation of differentiated CD8+T cells specific for respiratory viruses in the human lung. J. Exp. Med. 202, 1433–1442 (2005).
Piet, B. et al. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Invest. 121, 2254–2263 (2011).
Sathaliyawala, T. et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197 (2013).
Allen, R. J., Koutsakos, M., Hurt, A. C. & Kedzierska, K. Uncomplicated cystitis in an adult male following influenza B virus infection. Am. J. Case Rep. 18, 190–193 (2017).
Valkenburg, S. A. et al. Molecular basis for universal HLA-A*0201-restricted CD8+T-cell immunity against influenza viruses. Proc. Natl Acad. Sci. USA 113, 4440–4445 (2016).
Pascolo, S. et al. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J. Exp. Med. 185, 2043–2051 (1997).
Nguyen, T. H. et al. Understanding CD8(+) T-cell responses toward the native and alternate HLA-A*02:01-restricted WT1 epitope. Clin. Transl. Immunology 6, e134 (2017).
Epstein, S. L., Lo, C. Y., Misplon, J. A. & Bennink, J. R. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J. Immunol. 160, 322–327 (1998).
Babon, J. A., Cruz, J., Ennis, F. A., Yin, L. & Terajima, M. A human CD4+ T cell epitope in the influenza hemagglutinin is cross-reactive to influenza A virus subtypes and to influenza B virus. J. Virol. 86, 9233–9243 (2012).
Pedersen, S. R. et al. Immunogenicity of HLA class I and II double restricted influenza A-derived peptides. PLoS ONE 11, e0145629 (2016).
Tan, A. C. et al. The design and proof of concept for a CD8(+) T cell-based vaccine inducing cross-subtype protection against influenza A virus. Immunol. Cell Biol. 91, 96–104 (2013).
Wang, L. et al. Functional genomics reveals linkers critical for influenza virus polymerase. J. Virol. 90, 2938–2947 (2015).
Fulton, B. O., Sun, W., Heaton, N. S. & Palese, P. The influenza B virus hemagglutinin head domain is less tolerant to transposon mutagenesis than that of the influenza A virus. J. Virol. 92, e00754-18 (2018).
Allen, E. K. et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 23, 975–983 (2017).
Oshansky, C. M. et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care. Med. 189, 449–462 (2014).
Bird, N. L. et al. Oseltamivir prophylaxis reduces inflammation and facilitates establishment of cross-strain protective T cell memory to influenza viruses. PLoS ONE 10, e0129768 (2015).
Valkenburg, S. A. et al. Protective efficacy of cross-reactive CD8+ T cells recognising mutant viral epitopes depends on peptide-MHC-I structural interactions and T cell activation threshold. PLoS Pathog. 6, e1001039 (2010).
Bao, Y. et al. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82, 596–601 (2008).
Nguyen, T. H. et al. Maintenance of the EBV-specific CD8(+) TCRalphabeta repertoire in immunosuppressed lung transplant recipients. Immunol. Cell Biol. 95, 77–86 (2017).
Dudek, N. L. et al. Constitutive and inflammatory immunopeptidome of pancreatic beta-cells. Diabetes 61, 3018–3025 (2012).
Andreatta, M. & Nielsen, M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 (2016).
Lundegaard, C. et al. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 36, W509–W512 (2008).
Nielsen, M. et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 12, 1007–1017 (2003).
Colaert, N., Helsens, K., Martens, L., Vandekerckhove, J. & Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 (2009).
Eltahla, A. A. et al. Linking the T cell receptor to the single cell transcriptome in antigen-specific human T cells. Immunol. Cell Biol. 94, 604–611 (2016).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Acknowledgements
HLA-A2.1 transgenic HHD mice were developed by F. Lemonnier (Pasteur Institute). D227K and Q115E MHC-I constructs were developed and provided by L. Wooldridge (Bristol University). The authors acknowledge the provision of instrumentation, training and technical support by the Monash Biomedical Proteomics Facility. We thank all study participants and organ donors. We are grateful to the coordinators of DonateLife Victoria for obtaining organ tissues, and to L. Mariana and C. Kos (St. Vincent’s Institute) for their assistance. The Australian National Health and Medical Research Council (NHMRC) NHMRC Program Grant (1071916) to K.K. supported this work. M.K. is a recipient of a Melbourne International research scholarship and Melbourne International fee remission scholarship. S.S. is a recipient of a Victoria India doctoral scholarship and Melbourne International fee remission scholarship, University of Melbourne. K.K. is an NHMRC senior research level B fellow (1102792). J.R. is supported by an ARC laureate fellowship. S.G. is a Monash senior research fellow. F.L. and A.C.C. are NHMRC CDF2 fellows. E.B.C. and A.A.E. are NHMRC Peter Doherty fellows. The Melbourne WHO Collaborating Centre for Reference Research on Influenza is supported by the Australian Government Department of Health. A.W.P. is supported by an NHMRC principal research fellowship (1137739) and an NHMRC project grant (1085018) to A.W.P., T.C.K. and N.A.M. D.T. and D.V. are supported by contract HHSN272201400006C from the US National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services. P.T.I. was supported by a NHMRC Peter Doherty fellowship (1072159). P.G.T. is supported by the St. Jude Center of Excellence for Influenza Research and Surveillance (NIAID contract HHSN27220140006C), R01AI107625, R01AI136514, and American Lebanese Syrian Associated Charities. S.I.M. is supported by a JDRF Career Development award (5-CDA2014210-A-N) and NHMRC Project (GNT 1123586).
Author information
Authors and Affiliations
Contributions
M.K., K.K., T.H.O.N., P.T.I., N.A.M., A.W.P., S.G., D.V., F.L. and P.G.T. designed experiments. M.K., P.I., T.H.O.N., N.A.M., A.A.E., E.B.C., S.S., C.Y.W., B.Y.C., E.K.A., P.D., L.G., W.Z., D.F.B. and S.G. performed experiments. A.C.H., I.B., D.C.J., T.C.K., A.C.C., M.R., G.P.W., L.M.W., S.G.T., S.I.M., T.L., S.Rockman, M.E. and P.G.T. provided reagents and/or samples. M.K., P.I., T.H.O.N., J.C.C., S.S., S.Rizzetto, D.T., D.V., F.L. S.G., J.R., P.G.T., A.W.P. and K.K. analyzed data. M.K., T.H.O.N. and K.K. wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
S. Rockman is an employee of Seqirus but has no conflict of interest in the material presented. M.K., K.K. and E.B.C. are named as co-inventors in a patent application filed by the University of Melbourne (AU2017903652) covering the use of certain peptides described in the publication as part of vaccine formulation.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Integrated supplementary information
Supplementary Figure 1 Gating strategy for human CD8+ T cells for ICS assay.
CD8+ T cells were gated as CD8+CD3+CD14–CD19– live single lymphocytes.
Supplementary Figure 2 Immunopeptidomic analysis of human and viral peptides.
(a) Infection and viability rates for the immunopeptidomics experiments. (b) Experimental outline for immunopeptidomics analysis of IBV-infected C1R cells. Lysates were generated from C1R parental or C1R.A*02:01 cells and the transfected HLA-A*02:01, endogenous (parental) class I and class II molecules immunoaffinity purified sequentially using BB7.2 (HLA-A2 specific), w6/32 (pan class I) and a mixture of LB3.1 (HLA-DR), SPV-L3 (HLA-DQ) and B721 (HLA-DP). Peptides were eluted from the purified HLA and interrogated by LC–MS/MS using an information dependent acquisition (IDA) strategy and preliminary lists of HLA ligands generated for HLA-A*02:01, parental class I and class II. HLA-A*02:01 ligands were further refined by filtering for sequences in C1R parental purifications from all antibodies (represent non-specific pull down by BB7.2, and contamination with endogenous HLA molecules) as well as sequences in C1R.A*02:01 LB3.1/SPV-L3/B721 purifications (represent contamination with endogenous class II molecules). Peptides from C1R.A*02:01 w6/32 purifications were not included in the contaminant list due to the potential for excess HLA-A*02:01 not bound to the preceding BB7.2 resin to be isolated by w6/32. (c) Number of peptides identified per condition and experiment. (d) Distribution of IBV-derived HLA ligands across the B/Malaysia proteome, distinguished as likely parental class I (HLA-B*35:03 and HLA-C*04:01) or HLA class II ligands. All immunopeptidomic data are from two independent experiments.
Supplementary Figure 3 Dissection of peptide pools in human CD8+ T cell lines.
Frequency of IFN-γ+TNF+CD8+ T cells for individual peptides for each pool. Dots indicate individual donors and bars indicate the mean (n = 2–5). Red boxes indicate immunogenic peptides. (b) Frequency of responding donors for each immunogenic peptide (n = 6). (c, d) conservation of IBV peptides identified by immunopepidomics. (c) Conservation (average amino acid identity) of novel IBV peptides across IBV viruses in each lineage between 1940 and 2017. (d) Conservation (average amino acid identity) of novel IBV peptides in IAV and ICV. The bars indicate the percent conservation (average amino acid identity) of each peptide across the three types of viruses.
Supplementary Figure 4 Gating strategy for TAME and phenotyping of tetramer+ CD8+ T cells.
(a) Tetramer+ CD8+ T cells were identified within CD8+CD3+CD14–CD19– live single lymphocytes. (b) Representative FACS plots for each marker analyzed on tetramer+ cells. Total CD8+ cells are shown for comparison. (c) Representative FACS plots for Ki-67 staining on tetramer+ and total CD8+ cells along with an isotype control representative of one experiment.
Supplementary Figure 5 Phenotype of tetramer+ CD8+ T cells in healthy donors and during infection with influenza virus.
Expression of single markers on tetramer+ CD8+ T cells in healthy control (HC) and influenza-infected (IAV or IBV) patients or influenza-negative ILI patients (n = 3 for HC M158, 4 for PB1413 and BHA543 HC, 16 for IAV, 6 IBV, 7 Flu-neg ILI assessed over at least two independent experiments). Median and IQR are shown. A two tailed Mann-Whitney test was used to assess statistical significance. *p < 0.05, **p < 0.005.
Supplementary Figure 6 IBV infection of donor MK34 and expression of selected genes in tetramer+ CD8+ T cells from scRNA-seq.
(a) Phylogenetic analysis of PCR product from nasal swab. (b) Serological analysis before (~3 months prior) and after (~d14) IBV infection. Antibody titers against the two IBV lineages and IAV were assessed by haemagglutination inhibition assay. (c) Genes relating to T cell differentiation, activation and cytotoxicity or effector function were analyzed. Cells grouped by epitope and time point. (d) Validation of T cell activation genes from scRNA-seq by FACS. Violin plots show gene expression as determined by scRNA-seq and histograms show protein expression as determine by flow cytometry, for selected genes in available samples.
Supplementary Figure 7 Recall CD8+ T cell responses in HHD-A2 mice.
(a) Experimental outline for secondary infection with IBV. (b) Gating strategy. (c) Number of IFN-γ/TNF-producing CD8+ T cells specific for A2/BHA543 during 1o and 2o IBV infections (n = 5 mice for 1o and n = 4 for 2o). Mean and SEM are shown. (d) Polyfunctionality of BHA543 and BNS1266 during 1o and 2o IBV infections. % of triple (IFN-γ+IL-2+TNF+) CD8+ T cells of the two specificities during 1o and 2o IBV infections (n = 5 mice for 1o and n = 4 for 2o). Mean and SEM are shown. Statistical significance was determine using an unpaired two-tailed t-test, *p < 0.05, **p < 0.005.
Supplementary Figure 8 Lack of A2/PB1413 CD8+ T cells in HHD-A2 mice after infection with influenza virus or PB1413 vaccination.
Tetramer staining for A2/PB1413 after (a) primary, (b) secondary or (c) tertiary heterotypic infections. Response shown are from BAL, with similar results obtained from the spleen. A2/M158 and A2/BHA543 tetramer staining were used as positive controls. (d) Lack of A2/PB1413+CD8+ T cell responses after Pam2Cys lipopeptide vaccination. Splenocytes from vaccinated mice were enriched by TAME. An additional group of mice was vaccinated with an M158 lipopetide as a positive control. Representative FACS plots from a spleen are shown, with similar results obtained from the BAL. (e) Human PBMC stimulation with PB1413 lipopeptide to confirm its immunogenicity. (f) Lack of A2/PB1413 responses after peptide vaccination and IBV infection. Mice were vaccinated as in Supplementary Figure 7a. A2/BHA543 vaccination and tetramer staining were used as positive controls. (g) Lack of A2/PB1413-specific CD8+ T cells in naïve mice. Spleen and major lymph nodes were harvested and enriched by TAME. A C57/B6 mouse (A2–) was used as a control or background tetramer staining. TAME for A2/WT1A-specific CD8+ T cells was used as a positive control for naïve TAME. Data from one experiment.
Rights and permissions
About this article

Cite this article
Koutsakos, M., Illing, P.T., Nguyen, T.H.O. et al. Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat Immunol 20, 613–625 (2019). https://doi.org/10.1038/s41590-019-0320-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-019-0320-6
This article is cited by
-
Prevention of respiratory virus transmission by resident memory CD8+ T cells
Nature (2024)
-
Potential health and economic impact of paediatric vaccination using next-generation influenza vaccines in Kenya: a modelling study
BMC Medicine (2023)
-
In search of a pan-coronavirus vaccine: next-generation vaccine design and immune mechanisms
Cellular & Molecular Immunology (2023)
-
Using system biology and bioinformatics to identify the influences of COVID-19 co-infection with influenza virus on COPD
Functional & Integrative Genomics (2023)
-
Validation of Multi-epitope Peptides Encapsulated in PLGA Nanoparticles Against Influenza A Virus
Pharmaceutical Research (2023)
