
Abstract
Many diseases are driven by proteins that are aberrantly ubiquitinated and degraded. These diseases would be therapeutically benefited by targeted protein stabilization (TPS). Here we present deubiquitinase-targeting chimeras (DUBTACs), heterobifunctional small molecules consisting of a deubiquitinase recruiter linked to a protein-targeting ligand, to stabilize the levels of specific proteins degraded in a ubiquitin-dependent manner. Using chemoproteomic approaches, we discovered the covalent ligand EN523 that targets a non-catalytic allosteric cysteine C23 in the K48-ubiquitin-specific deubiquitinase OTUB1. We showed that a DUBTAC consisting of our EN523 OTUB1 recruiter linked to lumacaftor, a drug used to treat cystic fibrosis that binds ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR), robustly stabilized ΔF508-CFTR protein levels, leading to improved chloride channel conductance in human cystic fibrosis bronchial epithelial cells. We also demonstrated stabilization of the tumor suppressor kinase WEE1 in hepatoma cells. Our study showcases covalent chemoproteomic approaches to develop new induced proximity-based therapeutic modalities and introduces the DUBTAC platform for TPS.

Similar content being viewed by others

Main
Engaging the mostly undruggable proteome to uncover new disease therapies not only requires technological innovations that facilitate rapid discovery of ligandable hotspots across the proteome but also demands new therapeutic modalities that alter protein function through novel mechanisms1,2. Targeted protein degradation (TPD) tackles the undruggable proteome by targeting specific proteins for ubiquitination and proteasomal degradation. One major class of small-molecule effectors of TPD, proteolysis-targeting chimeras (PROTACs), are heterobifunctional molecules that consist of an E3 ligase recruiter linked to a protein-targeting ligand to induce the formation of ternary complexes that bring together an E3 ubiquitin ligase and the target protein as a neo-substrate3,4,5. PROTACs have enabled the targeted and specific degradation of numerous disease-causing proteins in cells3,6. New approaches for TPD have also arisen that exploit endosomal and lysosomal degradation pathways with lysosome-targeting chimeras or autophagy with autophagy-targeting chimeras7,8. New approaches for chemically induced proximity beyond degradation have also been developed in recent years, including targeted phosphorylation with phosphorylation-inducing chimeric small molecules and targeted dephosphorylation, but no small-molecule-based induced proximity approaches exist for targeted deubiquitination and subsequent stabilization of proteins9,10.
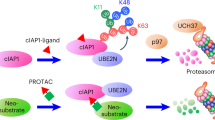
Active ubiquitination and degradation of proteins is the root cause of several classes of diseases, including many tumor suppressors in cancer (for example, TP53, CDKN1A, CDN1C and BAX), and mutated and misfolded proteins, such as ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) in cystic fibrosis or glucokinase in pancreatic cells in maturity-onset diabetes of the young type 2. In these cases, a TPS therapeutic strategy, rather than degradation, would be beneficial11,12,13,14. Analogous to TPD, we hypothesized that TPS could be enabled by the discovery of a small-molecule recruiter of a deubiquitinase (DUB) that could be linked to a protein-targeting ligand to form a chimeric molecule, which would induce the deubiquitination and stabilization of proteins of interest. We call this heterobifunctional stabilizer a DUBTAC (Fig. 1a). In this study, we report the discovery of a covalent recruiter for the K48-ubiquitin chain-specific DUB OTUB1, which when linked to a protein-targeting ligand stabilizes an actively degraded target protein to demonstrate proof of concept for the DUBTAC platform.

a, DUBTACs are heterobifunctional molecules consisting of a protein-targeting ligand linked to a DUB recruiter via a linker. DUBTACs are ideally used for stabilizing the levels of actively ubiquitinated proteins that are degraded by the proteasome. When treated in cells, a DUBTAC will induce the proximity of a DUB with a target protein to remove polyubiquitin chains to prevent the protein from undergoing proteasome-mediated degradation, thereby stabilizing and elevating the level of the actively degraded protein; Ub, ubiquitin. b, Of 65 DUBs mined in our research group’s aggregate chemoproteomic datasets of cysteine-reactive probe labeling with IA-alkyne in various complex proteomes, we identified probe-modified cysteines across 100% of the 65 DUBs. This is shown in the first pie chart. Among the 65 DUBs that showed probe-modified cysteines, 40 of these DUBs showed >10 aggregate spectral counts across our chemoproteomic datasets. Twenty-four, or 60%, of the 40 DUBs showed labeling of the DUB catalytic or active-site cysteines. c, Mining the DUB data, we identified 10 DUBs wherein there was one probe-modified cysteine that represented >50% of the total aggregate spectral counts for probe-modified cysteine peptides for the particular DUB. Seven of the 10 DUBs do not target a known catalytic cysteine, whereas three do target the catalytic cysteine (abbreviated ‘cat’); USP, ubiquitin-specific proteases; UCH, ubiquitin C-terminal hydrolases; OTU, ovarian tumor proteases. d, Analysis of aggregate chemoproteomic data for OTUB1 IA-alkyne labeling showing that C23 is the dominant site labeled by IA-alkyne compared with the catalytic (cat) C91. Chemoproteomic data analysis of DUBs across aggregated datasets can be found in Supplementary Table 1.
Results
Identifying allosteric ligandable sites within DUBs
To enable the DUBTAC platform, our first goal was to identify a small-molecule recruiter that targeted an allosteric site on a DUB without inhibiting DUB function, as the recruitment of a functional DUB would be required to deubiquitinate and stabilize the target protein. While many DUBs possess well-defined active sites bearing a catalytic and highly nucleophilic cysteine, there have not yet been systematic evaluations of allosteric, non-catalytic and ligandable sites on DUBs that could be pharmacologically targeted to develop a DUB recruiter. Chemoproteomic platforms, such as activity-based protein profiling (ABPP), have proven to be powerful approaches to map proteome-wide covalently ligandable sites. ABPP utilizes reactivity-based amino acid-specific chemical probes to profile reactive, functional and potentially ligandable sites directly in complex biological systems2,15. When used in a competitive manner, pretreatment with libraries of covalent small-molecule ligands can be used to screen for competition of probe binding to recombinant protein or complex proteomes to enable covalent ligand discovery against potential ligandable sites revealed by the reactivity-based probe2,16. Previous studies have shown that isotopic tandem orthogonal proteolysis-ABPP (isoTOP-ABPP) platforms for mapping sites of labeling with reactivity-based probes using quantitative proteomic approaches can identify hyperreactive, functional and ligandable cysteines15,16.
To identify DUB candidates that possess potential ligandable allosteric cysteines, we analyzed our research group’s aggregate chemoproteomic data of proteome-wide sites modified by reactivity-based probes collected since the start of our laboratory. Specifically, we mined our collective chemoproteomic data of cysteine-reactive alkyne-functionalized iodoacetamide (IA-alkyne) probe labeling sites from 455 isoTOP-ABPP experiments in human cell line proteomes for total aggregate spectral counts identified for each probe-modified site across the DUB family. We postulated that probe-modified cysteines within DUBs that showed the highest spectral counts aggregated over all chemoproteomic datasets compared to those sites within the same DUB that showed lower spectral counts may represent more reactive and potentially more ligandable cysteines. Caveats to this premise include cysteines that might be located in regions within a protein sequence that do not yield suitable tryptic peptides with respect to ionization and compatibility with MS-based sequencing and labeling of surface-exposed cysteines that may not be part of binding pockets. However, we conjectured that the aggregate chemoproteomics data would still yield candidate allosteric ligandable sites within DUBs that could be prioritized for covalent ligand screening. We initially mined our aggregate chemoproteomic data for 66 members of the cysteine protease family of DUBs, including ubiquitin-specific proteases, ubiquitin C-terminal hydrolases, Machado–Josephin domain proteases and ovarian tumor proteases (OTU), as they encompass the majority of DUB superfamilies. Interestingly, we found probe-modified cysteines across all of these DUB enzymes (Fig. 1b and Supplementary Table 1). Consistent with our aggregate chemoproteomic data of probe-modified sites being enriched in functional sites within DUBs, among the 40 DUBs that showed a total of >10 aggregate spectral counts of probe-modified peptides, 24 (60%) showed labeling of the DUB catalytic cysteine (Fig. 1b). We next prioritized this list of 40 DUBs to identify suitable candidates for TPS. We prioritized DUBs where the dominant probe-modified cysteine was (1) located at an allosteric site and not the catalytic cysteine such that we could target the identified cysteine with a covalent ligand while retaining the catalytic activity of the DUB; (2) in a dominantly identified probe-labeled peptide compared to other probe-modified sites within the same DUB, which could indicate a high degree of reactivity and potential covalent ligandability of the identified allosteric cysteine compared to the catalytic site and (3) frequently identified in chemoproteomics datasets, which would indicate the general accessibility of the cysteine in complex proteomes. We found 10 DUBs where one probe-modified cysteine represented >50% of the spectral counts of all modified cysteines for the particular protein, of which 7 of these DUBs showed primary sites of probe modification that did not correspond to the catalytic cysteine (Fig. 1c). Of these 10 DUBs, OTUB1 C23 was captured with >1,000 total aggregate spectral counts compared to <500 aggregate spectral counts for the other DUBs (Extended Data Fig. 1a). In our aggregated chemoproteomic data, the tryptic peptide encompassing OTUB1 C23 was the dominant peptide labeled by IA-alkyne, with >1,500 total spectral counts compared to 15 spectral counts for the peptide encompassing the catalytic C91 and 115 spectral counts for C212 (Fig. 1d).
OTUB1 is a highly expressed DUB that specifically cleaves K48-linked polyubiquitin chains, the type of ubiquitin linkage that destines proteins for proteasome-mediated degradation, and C23 represents a known ubiquitin substrate recognition site that is distant from the catalytic C91 (Extended Data Fig. 1b)17,18,19,20. Interestingly, C23 is also predicted to be in an intrinsically disordered region of the protein based on Predictor of Natural Disordered Regions (PONDR) analysis of OTUB1 (Extended Data Fig. 1b)21,22. Given our analysis of chemoproteomic data, the properties of OTUB1 and the location of C23, we chose OTUB1 as our candidate DUB for covalent ligand screening using gel-based ABPP approaches to discover an OTUB1 recruiter.
Discovering a covalent recruiter against OTUB1
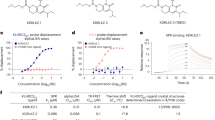
We performed a gel-based ABPP screen in which we screened 702 cysteine-reactive covalent ligands against labeling of pure OTUB1 protein with a rhodamine-functionalized cysteine-reactive iodoacetamide (IA-rhodamine) probe (Fig. 2a, Extended Data Fig. 1b and Supplementary Table 2). Through this screen, we identified the acrylamide EN523 (1) as a top hit (Fig. 2b). We confirmed that EN523 dose-responsively displaced IA-rhodamine labeling of OTUB1 without causing any protein aggregation or precipitation (Fig. 2c). We next performed liquid chromatography–tandem MS analysis (LC–MS/MS) of tryptic peptides from EN523 bound to OTUB1 and showed that EN523 selectively targets C23, with no detectable modification of the catalytic C91 (Fig. 2d). Following these data, we performed an in vitro reconstituted OTUB1 deubiquitination activity assay monitoring monoubiquitin release from diubiquitin and demonstrated that EN523 does not inhibit OTUB1 deubiquitination activity (Fig. 2e)23. These studies were performed in the presence of OTUB1-stimulating ubiquitin-conjugating enzyme E2 D1 (UBE2D1), an E2 ubiquitin ligase that engages in a complex with OTUB1 to stimulate OTUB1 activity.

a, Covalent ligand screen of a cysteine-reactive library competed against IA-rhodamine labeling of recombinant OTUB1 to identify binders to OTUB1 by gel-based ABPP. Vehicle DMSO or cysteine-reactive covalent ligands (50 μM) were preincubated with OTUB1 for 30 min at room temperature before IA-rhodamine labeling (500 nM for 30 min at room temperature). OTUB1 was then separated by SDS–PAGE, and in-gel fluorescence was assessed and quantified. Gel-based ABPP data and quantification of in-gel fluorescence are shown in Extended Data Fig. 1b and Supplementary Table 2. EN523 annotated in red was the top hit that showed the greatest inhibition of OTUB1 IA-rhodamine labeling. b, Structure of EN523, with cysteine-reactive acrylamide highlighted in red. c, Gel-based ABPP confirmation showing dose-responsive inhibition of IA-rhodamine binding of OTUB1. Vehicle DMSO or EN523 were preincubated with recombinant OTUB1 for 30 min at 37 °C before IA-rhodamine labeling (500 nM for 30 min at room temperature). OTUB1 was then separated by SDS–PAGE, and in-gel fluorescence was assessed. The silver staining demonstrates protein loading. Shown is a representative gel of n = 3 biologically independent samples per group. d, LC–MS/MS data showing an EN523-modified adduct on C23 of OTUB1. OTUB1 (10 μg) recombinant protein was incubated with EN523 (50 μM) for 30 min, after which the protein was precipitated and digested with trypsin, and tryptic digests were analyzed by LC–MS/MS to identify modified sites. e, OTUB1 DUB activity monitored by cleavage of K48 diubiquitin. Recombinant OTUB1 was preincubated with DMSO or EN523 (50 μM) for 1 h. After preincubation, OTUB1 was added to a mixture of diubiquitin and UBE2D1. The appearance of monoubiquitin (mono-Ub) was monitored by western blotting. f, Structure of the alkyne-functionalized EN523 probe NJH-2-075. g, Gel-based ABPP of NJH-2-075. Vehicle DMSO or NJH-2-075 were preincubated with OTUB1 for 30 min at 37 °C before IA-rhodamine labeling (500 nM for 30 min at room temperature). OTUB1 was then separated by SDS–PAGE, and in-gel fluorescence was assessed. Also shown is silver staining demonstrating protein loading. h, NJH-2-075 labeling of recombinant OTUB1. OTUB1 (0.5 μg) was labeled with DMSO or NJH-2-075 for 1.5 h at 37 °C, after which rhodamine-azide was appended by CuAAC, OTUB1 was separated by SDS–PAGE and in-gel fluorescence was assessed. Also shown is silver staining demonstrating protein loading. i, NJH-2-075 engagement of OTUB1 in HEK293T cells. HEK293T cells were treated with DMSO vehicle or NJH-2-075 (50 μM) for 2 h, after which cell lysates were subjected to CuAAC with biotin picolyl azide, and NJH-2-075-labeled proteins were subjected to avidin pulldown, elution, separation by SDS–PAGE and blotting for OTUB1 and vinculin. Both input lysate and pulldown levels are shown. Gels or blots shown in c, e and g–i are representative of n = 3 biologically independent samples per group. Raw gels and blots can be found in the source data.
We next used NMR analysis to further characterize EN523 binding to OTUB1. A 13C-heteronuclear multiple quantum coherence (HMQC) spectrum of OTUB1 revealed the presence of a homogenous and mostly folded protein with well-dispersed {U}-2H,1H/13C-methyl-Ile/Leu/Val/Ala (ILVA) methyl group resonances (Extended Data Fig. 2a). EN523 treatment of OTUB1 led to subtle but significant chemical shift perturbations (Extended Data Fig. 2b). As we did not assign any peaks in the OTUB1 spectrum, we could not determine the exact binding site of the small molecule. However, almost all affected resonances had stronger intensities than the average signal strengths and had chemical shifts that are close to the random coil values of the respective amino acids. These observations suggest that the amino acids giving rise to these resonances are located in unfolded sections of the protein (in agreement with our PONDR data), predicting that C23 (the site of EN523 binding) belongs to an intrinsically disordered region of the protein. Next, we investigated if the covalent binding of EN523 to C23 prevented the interaction of OTUB1 with the ubiquitin-loaded and free forms of UBE2D2. As the latter protein activates OTUB1 DUB activity, we wanted to confirm that EN523 did not interfere with this protein–protein interaction. We mixed OTUB1 with ubiquitinylated or free UBE2D2 and compared the chemical shifts of OTUB1 residues in the presence and absence of EN523. Binding of either form of the conjugating enzyme to OTUB1 induced strong peak perturbations (Extended Data Fig. 2c,d). These shift differences were almost identical for samples with and without EN523. The only differences we detected were shift changes for peaks that are affected by compound binding. These results indicated that EN523 did not interfere with binding of UBE2D2 to OTUB1.
We also explored structure–activity relationships (SARs) of our OTUB1 recruiter (2–12). Consistent with the necessity of the reactive acrylamide warhead for interacting with C23 of OTUB1, a non-reactive acetamide version of EN523, NJH-2-080 (2), showed loss in binding against OTUB1 (Extended Data Fig. 3). Replacing the methylfuran substituent with a benzoimidazolone, benzothiophene, benzofuran, phenyloxazole or imidazopyridine, but not methylimidazole, still retained potency against OTUB1 (Extended Data Fig. 3). We also explored preliminary SARs of the piperazinone core as well. Dimethyl and methylpiperazinone substitutions with a tert-butyl propionate extension from the furan still maintained potency against OTUB1. We also found that the (R)-methylpiperazinone derivative was more potent than the (S)-methylpiperazinone derivative of EN523, indicating that we may be able to achieve stereochemically specific interactions with OTUB1 (Extended Data Fig. 3). These data also demonstrated that extension off the furan may present an optimal exit vector for synthesis of DUBTACs. Overall, while the SAR showed room for flexibility within the EN523 core scaffold, we did not identify significantly more potent OTUB1 ligands, and thus we chose to further pursue EN523 as our OTUB1 recruiter for follow-up studies.
An alkyne-functionalized probe of EN523 (NJH-2-075 (13)) was then synthesized with the goal of assessing whether this ligand engaged OTUB1 in cells (Fig. 2f). NJH-2-075 retained binding to OTUB1 in vitro, as shown by (1) gel-based ABPP demonstrating competition of NJH-2-075 against IA-rhodamine labeling of recombinant OTUB1 and (2) direct labeling of recombinant OTUB1 by NJH-2-075 visualized by copper-catalyzed azide-alkyne cycloaddition (CuAAC) of azide-functionalized rhodamine to NJH-2-075-labeled OTUB1 (monitored by in-gel fluorescence; Fig. 2g,h). We demonstrated NJH-2-075 engagement of OTUB1 in cells by enrichment of endogenous OTUB1, but not that of an unrelated protein vinculin, through NJH-2-075 compared to vehicle treatment in HEK293T cells (Fig. 2i). Collectively, these data highlighted EN523 as a promising covalent OTUB1 ligand that targeted a non-catalytic and allosteric C23 on OTUB1 without inhibiting OTUB1 deubiquitination activity and engaged OTUB1 in cells.
Showing proof of concept of DUBTACs with mutant CFTR
To demonstrate the feasibility of using EN523 as an OTUB1-recruiting module of a heterobifunctional DUBTAC, we identified the mutant ΔF508-CFTR chloride channel as a proof-of-concept case where protein stabilization would be therapeutically desirable. ΔF508, a frameshift mutation caused by deletion at codon 508 in exon 10 of CFTR, resulting in the absence of a phenylalanine residue, is the most common mutation that induces the cystic fibrosis phenotype24. This mutation causes the protein to become conformationally unstable, leading to K48 polyubitiquination and degradation before trafficking from the endoplasmic reticulum to the cell surface14,24. Previous studies have demonstrated the feasibility of stabilizing mutant CFTR not only by genetic and pharmacological inhibition of the cognate E3 ligase RNF5 but also through targeted recruitment of DUBs using a genetically encoded and engineered DUB targeted to CFTR using a CFTR-targeting nanobody25,26,27. Importantly for our work, suitable CFTR-targeting small-molecule ligands exist. Lumacaftor, a drug for cystic fibrosis developed by Vertex Pharmaceuticals, acts as a chemical chaperone for ΔF508-CFTR and corrects its misfolding, leading to increased trafficking of ΔF508-CFTR to the cell membrane and partial restoration of protein function28. Despite lumacaftor’s chaperoning activity, the vast majority of ΔF508-CFTR is still actively ubiquitinated and degraded, making the potential of a synergistic effect via DUBTAC-induced deubiquitination a therapeutically attractive option.
With this in mind, we synthesized DUBTACs linking the OTUB1 recruiter EN523 to the CFTR chaperone lumacaftor with two different C3 or C5 alkyl linkers, NJH-2-056 (14) and NJH-2-057 (15) (Fig. 3a,b). We confirmed that these two DUBTACs still engaged recombinant OTUB1 in vitro by gel-based ABPP (Fig. 3c,d). We used CFBE41o-4.7 human bronchial epithelial cells expressing ΔF508-CFTR, a human cystic fibrosis bronchial epithelial cell line, as a model system to test our DUBTACs. We first showed that EN523 did not alter OTUB1 protein levels in these cells (Extended Data Fig. 4a). We also demonstrated that the alkyne-functionalized EN523 probe NJH-2-075 still engaged OTUB1 in this cell line (Extended Data Fig. 4b). The DUBTACs were then tested in these cells alongside lumacaftor or EN523 treatment alone. Treatment with NJH-2-056, lumacaftor, or EN523 did not alter mutant CFTR levels; however, we observed a robust and significant increase in CFTR protein levels with NJH-2-057 treatment (Fig. 3e,f). This stabilization was dose responsive and time dependent (Extended Data Fig. 5). We further confirmed that the stabilized protein was CFTR using three additional commercially available CFTR antibodies (Extended Data Fig. 6) and showed that the DUBTAC-stabilized CFTR band was attenuated following CFTR knockdown (Extended Data Fig. 7).

a,b, Structures of NJH-2-056 (a) and NJH-2-057 (b); these DUBTACs against mutant CFTR protein are formed by linking the CFTR ligand lumacaftor to the OTUB1 recruiter EN523 through C3 and C5 alkyl linkers, respectively. c,d, Gel-based ABPP analysis of NJH-2-056 (c) and NJH-2-057 (d) against OTUB1. Vehicle DMSO or DUBTACs were preincubated with recombinant OTUB1 for 30 min at 37 °C before addition of IA-rhodamine (100 nM) for 30 min at room temperature. OTUB1 was run on SDS–PAGE, and in-gel fluorescence was assessed. Protein loading was assessed by silver staining. e, Effect of DUBTACs on mutant CFTR levels. CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO, NJH-2-056 (10 μM), NJH-2-057 (10 μM), lumacaftor (10 μM) or EN523 (10 μM) for 24 h, and mutant CFTR and loading control GAPDH levels were assessed by western blotting. f, Quantification of the experiment described in e. g, TMT-based quantitative proteomic profiling of NJH-2-057 treatment. CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 (10 μM) for 24 h. Data shown are from n = 3 biologically independent samples per group. Full data for this experiment can be found in Supplementary Table 3. h, Native MS analysis of DUBTAC-mediated ternary complex formation. OTUB1 (2 μM) and the CFTR nucleotide-binding domain 1 (2 μM) were incubated with DMSO vehicle, EN523 (50 μM) or NJH-2-057 (50 μM) in 150 mM ammonium acetate with MgCl2 (100 μM) and ATP (100 μM). Representative mass spectra from n = 3 biologically independent samples per group are shown. i, Percentage of ternary complex formation assessed by measuring the CFTR–OTUB1 complex formed in the experiment described in h. Gels shown in c, d and e are representative of n = 3 biologically independent samples per group. Data in f and i show individual biological replicate values and average ± s.e.m. from n = 3 biologically independent samples per group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests in f and i compared with vehicle-treated controls and is expressed as *P < 0.05. Raw gels and blots, bar graph data and exact P values can be found in the source data.
We also explored the dependence of CFTR stabilization on linker length and composition (14–20) (Extended Data Fig. 8). DUBTACs bearing C5 and C6 alkyl linkers, but not C3 and C4 alkyl linkers, stabilized CFTR. Interestingly, none of the DUBTACs bearing PEG linkers were able to stabilize CFTR (Extended Data Fig. 8). We also made eight additional CFTR DUBTACs bearing more rigid heterocycle-containing linkers (21–28) to determine whether these compounds that may be less flexible and potentially more drug-like may perform better in stabilizing mutant CFTR (Extended Data Fig. 9). Interestingly, nearly all these DUBTACs increased CFTR levels, with substantially improved response from GL-03 (23) (bearing a fused azepane-pyrrolidine linker) compared to NJH-2-075 (Extended Data Fig. 9). Consistent with the necessity of the cysteine-reactive warhead in binding to OTUB1, we also demonstrated that a non-reactive propionamide version of NJH-2-057, NJH-2-106 (29), was incapable of stabilizing CFTR (Extended Data Fig. 10). The alkyne probe NJH-2-075, which contains an identical linker to NJH-2-057 but exchanges an ethynylphenyl group for lumacaftor, also did not induce CFTR stabilization (Extended Data Fig. 10). The CFTR smear that we observed in the blot with NJH-2-057 treatment is consistent with previous studies investigating CFTR and in line with our observations with the proteasome inhibitor bortezomib that would maximally stabilize actively ubiquitinated and degraded mutant CFTR protein (Supplementary Fig. 1). This smear likely represents a combination of differential glycosylation states, other forms of ubiquitination on CFTR that may not be removed by OTUB1 (for example, K63 ubiquitin chains) and previously observed anomalous migration of CFTR on SDS–PAGE due to the presence of SDS-resistant ternary structures within the protein24,29. Based on the molecular weight of the darkest part of the CFTR blot >225 kDa, we conjectured that we are stabilizing the fully mature glycosylated form of mutant CFTR (Fig. 3e).
To further validate our western blot data for CFTR stabilization and to assess the proteome-wide activity of NJH-2-057, we performed a tandem mass tags (TMT)-based quantitative proteomic analysis of NJH-2-057-treated CFBE41o-4.7 cells expressing ΔF508-CFTR. Satisfyingly, the proteomic analysis showed CFTR among the most robustly stabilized proteins (ratio of 7.8 comparing NJH-2-057 to vehicle treatment; Fig. 3g and Supplementary Table 3). While there were additional proteins with significant changes in abundance levels, we only observed 21 proteins that were significantly stabilized by greater than fivefold with an adjusted P value of <0.01 compared to vehicle-treated controls out of 4,552 total quantified proteins (Fig. 3g and Supplementary Table 3). These observed changes in protein abundance levels by the DUBTAC appear to be DUBTAC-specific changes because these changes were not detected with EN523 or lumacaftor treatment alone (Supplementary Fig. 2 and Supplementary Table 3) and may represent compensatory changes occurring from elevations in CFTR levels in cells or could represent changes resulting from off-targets of the DUBTAC. Interestingly, among proteins elevated along with CFTR were several protein chaperones, including heat shock 70-kDa protein 6, DnaJ heat shock protein family Hsp40 member B1, heat shock 50-kDa protein 1A and DNAJ homolog subfamily B member 4. These changes could reflect potential compensatory on-target upregulation of protein chaperones in response to highly elevated levels of a relatively unstable mutant CFTR. Nonetheless, we did not observe widespread alterations in protein levels with DUBTAC treatment, suggesting that we were not substantially disrupting global protein turnover.
Having identified NJH-2-057 as a DUBTAC that was capable of stabilizing mutant CFTR in cells, we next sought to confirm the formation of a ternary complex between CFTR, NJH-2-057 and OTUB1 in vitro using recombinant protein and native MS-based approaches (Fig. 3h,i). While the highest intensity signals corresponded to unmodified OTUB1 and the ΔF508-harboring CFTR nucleotide-binding domain used in this experiment, potentially indicating low levels of target engagement under these experimental conditions, we observed significant CFTR–OTUB1 complex formation with NJH-2-057 treatment but not with DMSO vehicle or EN523 treatment (Fig. 3h,i). The predominantly observed mass for this complex corresponded to OTUB1 and CFTR but not the combined masses of OTUB1, CFTR and NJH-2-057. This may be because the NJH-2-057 adduct on OTUB1 may be unstable to electrospray ionization and desolvation energy conditions required to observe the protein complex, as we were also not able to observe the NJH-2-057 mass adduct on OTUB1. However, minor peaks indicated the presence of adducts consistent with either full NJH-2-057 or either MS-induced fragments or breakdown products (Supplementary Fig. 3). Nonetheless, given that this OTUB1–CFTR complex was only observed with DUBTAC treatment but not with DMSO or EN523 treatment, our data strongly suggest that the DUBTAC enables ternary complex formation.
To further confirm that the robust stabilization in mutant CFTR levels conferred by NJH-2-057 was due to the proposed on-target activity, we demonstrated that stabilization of CFTR was attenuated by pretreatment with either lumacaftor or EN523, indicating that stabilization by the DUBTAC was due to targets engaged by both lumacaftor and EN523 (Fig. 4a,b). These data also indicate the necessity for ternary complex formation to stabilize CFTR levels. To further verify that the CFTR stabilization was dependent on OTUB1, OTUB1 knockdown significantly attenuated mutant CFTR stabilization by NJH-2-057 (Fig. 4c,d).

a, Effect of lumacaftor or EN523 preincubation on NJH-2-057 DUBTAC-mediated stabilization of mutant CFTR levels. CFBE41o-4.7 cells expressing ΔF508-CFTR were pretreated with vehicle DMSO, lumacaftor (100 μM) or EN523 (100 μM) for 1 h before treatment with NJH-2-057 (10 μM) for 24 h. Mutant CFTR and loading control GAPDH levels were assessed by western blotting. b, Quantification of the experiment described in a. c, Effect of OTUB1 knockdown on NJH-2-057 DUBTAC-mediated mutant CFTR stabilization. CFBE41o-4.7 cells expressing ΔF508-CFTR were transiently transfected with non-targeting short interfering RNA (siRNA; siControl) or siOTUB1 oligonucleotides for 48 h before treatment of cells with vehicle DMSO or NJH-2-057 (10 μM) for 16 h. Mutant CFTR, OTUB1 and loading control GAPDH levels were assessed by western blotting. d, Levels of mutant CFTR and OTUB1 from the experiment described in c. e, Transepithelial conductance, expressed as short-circuit current (Isc), in primary human cystic fibrosis donor bronchial epithelial cells bearing the ΔF508-CFTR mutation. Cells were treated with DMSO vehicle, NJH-2-057 (10 μM) or lumacaftor (10 μM) 24 h before the TECC24 assay in which cells received four additional sequential treatments with a sodium channel inhibitor amiloride (10 μM), a cAMP activator forskolin (20 μM), a CFTR potentiator VX770 (0.5 μM) and a CFTR inhibitor CFTR(inh)-172 (30 μM). Shown are the average values from conductance from a single donor. Experiments were conducted in primary cells from two donors. f, Changes in current between potentiator VX770 (ivacaftor) treatment and the CFTR inhibitor treatment in the experiment described in e in two primary human cystic fibrosis donor bronchial epithelial cells bearing the ΔF508-CFTR mutation. Individual replicate data are shown in the bar graph from n = 10 biologically independent samples in the DMSO vehicle-treated group, n = 13 biologically independent samples in the lumacaftor-treated group and n = 18 biologically independent samples in the NJH-2-057-treated group. Gels shown in a and c are representative of n = 3 biologically independent samples per group. Data in b and d show individual biological replicate values and average ± s.e.m. from n = 3 biologically independent samples per group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests in b, d and f and is expressed as *P < 0.05 compared with vehicle-treated control in b and f and compared with vehicle-treated siControl in d. Statistical significance is expressed as #P< 0.05 compared with the NJH-2-057-treated group in b and compared with the NJH-2-057-treated siControl group for CFTR levels in d. Raw gels and blots, bar graph and line plot data and exact P values can be found in the source data.
We next performed a competitive isoTOP-ABPP study to assess the overall proteome-wide selectivity and cysteine reactivity of NJH-2-057 treatment in CFBE41o-4.7 cells expressing ΔF508-CFTR (Supplementary Fig. 4 and Supplementary Table 4). Of 1,270 IA-alkyne probe-modified peptides quantified in two of three biological replicates, there were only five targets that showed isotopically light-to-heavy or control-to-NJH-2-057 treatment probe-modified peptide ratios of >4 with an adjusted P value of <0.05: VDAC2 C76, TUBB1 C201, RLF C744, VDAC2 C47 and VDAC3 C66 (Supplementary Fig. 4 and Supplementary Table 4). Yang et al. previously showed that Nedd4 could ubiquitinate VDAC2 and VDAC3, and overexpression of Nedd4 significantly increased K48-linked ubiquitination of VDAC2 and VDAC3 (ref. 30). However, none of these targets would be expected to directly influence the activity of our DUBTAC. OTUB1 C23 was captured in our isoTOP-ABPP experiment but only showed a ratio of 1.6, which would correspond to ~60% target occupancy (Supplementary Table 3). This likely indicates that the observed CFTR stabilization by NJH-2-057 is occurring through relatively low levels of OTUB1 occupancy in cells, which would also be in line with our in vitro labeling and native MS data. The activity of heterobifunctional molecules has also been reported previously in studies using covalent E3 ligase recruiters for targeted protein degradation applications showing that relatively minimal target occupancy of E3 ligases can still lead to robust degradation of target proteins due to the catalytic mechanism of action of the E3 ligases31,32,33,34. We conjectured that a similar catalytic effect in a DUBTAC also leads to robust stabilization of the target protein with partial OTUB1 occupancy.
Having shown stabilization of CFTR protein levels with our DUBTAC, we next sought to determine whether our DUBTAC-mediated increase in CFTR protein levels led to improved cell surface CFTR function (Fig. 4e,f). We measured transepithelial conductance in primary human cystic fibrosis donor bronchial epithelial cells bearing the ΔF508-CFTR mutation. These cells were pretreated with vehicle, lumacaftor or NJH-2-057 for 24 h before sequential treatments with a sodium channel inhibitor amiloride, a cAMP activator forskolin and a CFTR potentiator VX770 (ivacaftor) to fully activate CFTR function in cells. After chloride channel conductance was potentiated with VX770, the cells were treated with a CFTR inhibitor, CFTR(inh)-172, to show CFTR dependence of any increases observed in transepithelial conductance. The difference in conductance between potentiator VX770 and CFTR inhibitor treatment were quantified under the three different treatment conditions (vehicle, lumacaftor or DUBTAC treatment) to ascertain the effects that our DUBTAC had on CFTR-mediated conductance compared to lumacaftor or vehicle treatment (Fig. 4e,f). Our studies showed that treatment of these primary cells with NJH-2-057 led to significant improvement in CFTR-dependent transepithelial conductance compared to lumacaftor or vehicle treatment, indicating that our CFTR DUBTAC elevated not only CFTR protein levels but also functional CFTR at the cell surface, leading to improved CFTR function (Fig. 4e,f).
Using DUBTACs to stabilize WEE1
We next sought to show a second example of TPS with DUBTACs against another actively degraded target for which a well-validated ligand had been reported in the literature. We selected WEE1, a tumor suppressor kinase in non-malignant eukaryotic somatic cells that phosphorylates the cyclin-dependent kinase (CDK1)–cyclin B1 complex to inhibit cell cycle progression during S and G2 phases of mitosis and whose activity must be downregulated for mitotic progression to occur35,36. One mechanism through which WEE1 activity is suppressed is via ubiquitin-mediated proteasomal degradation36,37. Clinical WEE1 inhibitors, such as AZD1775, have been developed to be given in combination with DNA-damaging chemotherapy agents for inducing premature mitosis to exert anticancer effects, and WEE1 PROTACs using AZD1775 have also been developed to selectively degrade WEE1 in cancer cells38. We first confirmed previously reported results that treatment of HEP3B hepatoma cancer cell lines with a proteasome inhibitor bortezomib stabilized WEE1 levels, demonstrating that WEE1 was regulated by ubiquitin-mediated proteasomal degradation in this cell line (Fig. 5a)37. We next synthesized four DUBTACs linking AZD1775 to our OTUB1 recruiter EN523 through no linker (LEB-03-153) (30), a C3 alkyl linker (LEB-03-144) (31), a C5 alkyl linker (LEB-03-145) (32) or a PEG2 linker (LEB-03-146) (33) (Fig. 5b). LEB-03-144 and LEB-03-146 with the C3 alkyl linker and the PEG2 linker, respectively, showed significant WEE1 stabilization in HEP3B cells comparable to WEE1 levels observed with bortezomib treatment, whereas EN523 or AZD1775 treatment alone had no impact on WEE1 levels (Fig. 5c,d). While the therapeutic relevance of a WEE1 DUBTAC using a WEE1 inhibitor remains to be seen, these data show additional mechanistic proof of concept for TPS using DUBTACs.

a, HEP3B cells were treated with DMSO vehicle or bortezomib (1 μM) for 24 h. WEE1 and loading control GAPDH levels were assessed by western blotting. b, Structures of four WEE1 DUBTACs linking AZD1775 to the OTUB1 recruiter EN523 through four different linkers. c, HEP3B cells were treated with DMSO vehicle, the four DUBTACs, bortezomib, EN523 or AZD1775 at 1 μM for 24 h. WEE1 and loading control GAPDH levels were assessed by western blotting. d, Quantitation of the data shown in c. Blots shown in a and b are representative blots from n = 3 biologically independent samples per group. Data in bar graphs show individual biological replicate values and average ± s.e.m. from n = 3 biologically independent samples per group. Raw blots, bar graphs and exact P values can be found in the source data.
Discussion
In this study, we discovered a covalent small-molecule recruiter EN523 for the K48-ubiquitin chain-specific DUB OTUB1. We demonstrated that this recruiter can be incorporated into fully synthetic heterobifunctional DUBTACs by linking a DUB recruiter to protein-targeting ligands to enable TPS of actively degraded target proteins in cells. We showed two successful examples of TPS with ΔF508-CFTR and WEE1. For ΔF508-CFTR, we also demonstrated that we not only heightened the levels of the mutant protein but also improved cell surface chloride channel conductance of CFTR with our DUBTAC in combination with the potentiator ivacaftor compared to lumacaftor and ivacaftor treatments. While we showed early validation of the DUBTAC platform here, there are many avenues for future exploration. These include further optimization of DUB recruiters against OTUB1 to improve their potency and proteome-wide selectivity as well as the discovery of new recruiters against other candidate DUBs. Much like with PROTACs, we observe significant dependency on linker length and composition for stabilizing target proteins with DUBTACs. Further exploration of this linker dependence with DUBTACs will be necessary for improving potency and kinetics of stabilization and bioavailability. In addition, elucidating the mechanism, structural underpinnings and kinetics in the formation of the ternary complex formed between the target protein and DUB and understanding how the target protein is deubiquitinated by the DUBTAC will be important. Using more advanced methods beyond native MS to monitor ternary complex formation, including AlphaLISA and time-resolved fluorescence resonance energy transfer, will be useful to achieve these goals39.
Given our initial proof of concept for CFTR and WEE1 stabilization with a DUBTAC, there are many promising areas that could benefit from targeted deubiquitination of actively ubiquitinated and degraded proteins to provide therapeutic benefit. Targets that could benefit from a DUBTAC that already possess protein-targeting ligands include stabilizing BAX levels in the mitochondria to induce apoptosis, stabilizing STING for immunooncology applications or stabilizing glucokinase in pancreatic cells for maturity-onset diabetes of the young type 2 (refs. 40,41,42,43). Other targets that would benefit from a DUBTAC would be various tumor suppressors that are actively ubiquitinated and degraded to maintain cancer cell proliferation44. There are also many other genetic disorders beyond cystic fibrosis where mutations can lead to protein destabilization and ubiquitin-mediated degradation that can also be stabilized by DUBTACs for therapeutic benefit. These include glucocerebrosidase mutations in Gaucher’s disease or Parkinson’s disease45 and phenylalanine hydroxylase and fumarylacetoacetate hydroxylase mutations in phenylketonuria46. These disorders could directly benefit from TPS via DUBTACs. In diseases caused by haploinsufficiency where loss of one copy of a gene leads to disease pathology, DUBTACs could potentially slow down the turnover rate of the protein to increase the levels of the protein to alleviate the disease47. Overall, our study puts forth the discovery of DUB recruiters and shows proof of concept for the DUBTAC platform for TPS via induced proximity of a DUB with a target protein. In addition, our study underscores the utility of using chemoproteomics-enabled covalent ligand discovery platforms to facilitate development of unique induced proximity-based therapeutic modalities beyond TPD.
Methods
Materials
Cysteine-reactive covalent ligand libraries were either previously synthesized and described or, for the compounds starting with ‘EN’, were purchased from Enamine, including EN523 (refs. 31,34,48,49,50,51). Lumacaftor was purchased from MedChemExpress.
Cell culture
CFBE41o-4.7 ΔF508-CFTR human cystic fibrosis bronchial epithelial cells were purchased from Millipore Sigma (SCC159). CFBE41o-4.7 ΔF508-CFTR human cystic fibrosis bronchial epithelial cells were cultured in MEM (Gibco) containing 10% (vol/vol) FBS and maintained at 37 °C with 5% CO2. Hep3B2.1-7 (HEP3B) cells were purchased from ATCC (HB-8064) and cultured in EMEM (ATCC) containing 10% (vol/vol) FBS and maintained at 37 °C with 5% CO2.
Gel-based activity-based protein profiling
Recombinant OTUB1 (0.1 μg per sample) was pretreated with either DMSO vehicle or covalent ligand or DUBTACs at 37 °C for 30 min in 25 μl of PBS and subsequently treated with IA-rhodamine (concentrations are designated in the figure legends; Setareh Biotech) at room temperature for 1 h. The reaction was stopped by addition of 4× reducing Laemmli SDS sample loading buffer (Alfa Aesar). After boiling at 95 °C for 5 min, the samples were separated on precast 4–20% Criterion TGX gels (Bio-Rad). Probe-labeled proteins were analyzed by in-gel fluorescence using a ChemiDoc MP (Bio-Rad).
NJH-2-057 probe labeling of recombinant OTUB1
Recombinant and pure OTUB1 protein (0.5 μg) per sample per replicate was suspended in 50 μl of PBS. One microliter of either DMSO or NJH-2-075 (to give final concentrations of 50, 10, 1 and 0.1 μM) was added, followed by a 1.5-h incubation at 37 °C. Next, 7.8 μl of a solution composed of 9.4 μl of 5 mM azide-Fluor 545 (in DMSO), 112 μl of tris(benzyltriazolylmethyl)amine (TBTA) ligand (stock 1.7 mM in four parts tert-butanol + one part DMSO), 37.5 μl of 50 mM TCEP (in water) and 37.5 μl of 50 mM copper(II) sulfate was added to each sample, and the samples were incubated for 1 h at room temperature. Following CuAAC, 30 μl of Laemmli sample buffer (4×) was added to each sample, vortexed and boiled for 6 min at 95 °C. Samples were loaded on an SDS–PAGE gel and analyzed for in-gel fluorescence.
Deubiquitinase activity assay
Previously described methods were used to assess EN523 effects on OTUB1 activity23. Recombinant OTUB1 (500 nM) was preincubated with DMSO or EN523 (50 μM) for 1 h. To initiate the assay, pretreated OTUB1 enzyme was mixed 1:1 with diubiquitin reaction mix for final concentrations of 250 nM OTUB1, 1.5 µM diubiquitin, 12.5 µM UBE2D1 and 5 mM DTT. The appearance of monoubiquitin was monitored by western blotting over time by removing a portion of the reaction mix and adding Laemmli’s buffer to terminate the reaction. The blot shown is a representative gel from n = 3 biologically independent experiments per group.
Bio-nuclear magnetic resonance analysis of EN523–OTUB1 interactions
We recorded all NMR spectra on a Bruker 600-MHz spectrometer equipped with a 5-mm QCI-F cryo probe with a z gradient, and the temperature was kept constant at 298 K during all experiments. To probe compound and E2 ligase binding to OTUB1, we recorded 1H-1D and 13C-SOFAST-HMQC experiments. We used 3-mm NMR tubes filled with 160 µl of 50 µM ILVA, {U}-15N labeled OTUB1, 25 mM d-Tris, pH 7.5, 150 mM NaCl, 5% heavy water (to lock), 100 µM sodium trimethylsilylpropanesulfonate (DSS; internal standard), 75 µM EN523 (dissolved in 100% d6-DMSO; for compound-binding study) and/or 100 µM E2 D2/UBE2D2 (for ligase-binding studies). To allow for complete binding of the compound to OTUB1, we chose an incubation period of ~40 h. We also recorded reference spectra with the adequate volumes of pure d6-DMSO and/or E2 buffer to compensate for solvent-induced effects and repeated experiments after 40 h to make sure that any spectral changes were not related to protein oxidation.
Labeling of endogenous OTUB1 in HEK293T cells with NJH-2-075 probe
One plate of 70% confluent HEK293T cells per condition per replicate were treated with either DMSO vehicle or NJH-02-075 (50 μM) for 2 h. Cells were collected by scraping, suspended in 600 μl of PBS, lysed by probe sonication and centrifuged for 10 min at 5,000 r.p.m. to remove debris. Lysate was normalized to 3.1 mg ml–1, and 85 μl was removed for western blotting analysis of input. Lysate (500 μl) was then incubated for 1 h at room temperature with 10 μl of 5 mM biotin picolyl azide (in water), 10 μl of 50 mM TCEP (in water), 30 μl of TBTA ligand (stock 1.7 mM in four parts tert-butanol + one part DMSO) and 10 μl of 50 mM copper(II) sulfate. Following CuAAC, precipitated proteins were washed three times with cold methanol and resolubilized in 200 μl of 1.2% SDS/PBS. To ensure solubility, proteins were heated to 90 °C for 5 min following resuspension. PBS (1 ml) was then added to each sample, followed by 50 μl of high-capacity streptavidin beads. Samples were then incubated overnight on a rocker at 4 °C. The following morning, the samples were warmed to room temperature, and non-specific binding proteins were washed away with three PBS washes followed by three water washes. Beads were then resuspended in 100 μl of PBS and 30 μl of Laemmli sample buffer (4×) and boiled for 13 min at 95 °C. Samples were vortexed and loaded onto an SDS–PAGE gel along with saved input samples for western blotting analysis.
Western blotting
Proteins were resolved by SDS–PAGE and transferred to nitrocellulose membranes using the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked with 5% BSA in Tris-buffered saline containing Tween 20 (TBS-T) solution for 30 min at room temperature, washed in TBS-T and probed with primary antibody diluted in the recommended diluent, per the manufacturer’s instructions, overnight at 4 °C. After three washes with TBS-T, the membranes were incubated in the dark with IR680- or IR800-conjugated secondary antibodies at a 1:10,000 dilution in 5% BSA in TBS-T at room temperature for 1 h. After three additional washes with TBS-T, blots were visualized using an Odyssey Li-Cor fluorescent scanner. The membranes were stripped using ReBlot Plus Strong Antibody Stripping Solution (EMD Millipore) when additional primary antibody incubations were performed. Antibodies used in this study were CFTR (Cell Signaling Technologies, rabbit monoclonal antibody 78335, 1:1,000 dilution in 5% BSA; Figs. 3 and 4), CFTR (R&D Systems, mouse monoclonal antibody, MAB25031, 1 μg ml–1 in 5% BSA; Extended Data Fig. 3), CFTR (Millipore, mouse monoclonal antibody, MAB3484, 1 μg ml–1 in 5% BSA; Extended Data Fig. 3), CFTR (Prestige, rabbit polyclonal antibody, HPA021939, 1:1,000 dilution in 5% BSA; Extended Data Fig. 3), GAPDH (Proteintech, mouse monoclonal antibody, 1:10,000 dilution in 5% BSA, 60004-1-Ig), OTUB1 (Abcam, rabbit monoclonal antibody, ab175200, 1:1,000 dilution in 5% BSA, EPR13028(B)), CTNNB1 (Cell Signaling Technologies, 1:1,000 dilution in 5% BSA, rabbit monoclonal antibody, 8480) and WEE1 (Cell Signaling Technologies, 1:1,000 dilution in 5% BSA, 4936).
Native mass spectrometry analysis of ternary complex formation
Native MS experiments were performed on a Thermo QE UHMR equipped with a nano-electrospray ionization source (Advion TriVersa NanoMate). Recombinant OTUB1 was first buffer exchanged into 150 mM ammonium acetate, 100 µM MgCl2 and 100 µM ATP at pH 6.7. OTUB1 (4 µM) was then preincubated at room temperature for 24 h with DMSO, EN523 (100 µM) or NJH-2-057 (100 µM). After 24 h, 4 µM CFTRΔ (NBD1 domain-amino acids 387-646, Δ405-436; Δ508), in the same buffer, was added to the OTUB1 solution, for final concentrations of 2 µM of each protein with either DMSO or 50 µM compound. The solution was then allowed to incubate for 30 min before analysis on the mass spectrometer. Mass spectra were recorded in positive ion mode with a mass range of 1,000–8,000 m/z. Each spectrum was then deconvoluted, and relevant peaks were integrated to determine percent ternary complex formed. All experiments were performed in triplicate.
Isotopic tandem orthogonal proteolysis activity-based protein profiling chemoproteomic experiments
IsoTOP-ABPP studies were done as previously reported15,31,52. Our aggregate chemoproteomic data analysis of DUBs was obtained from 455 distinct isoTOP-ABPP experiments performed by the Nomura Research Group. These data are aggregated from various human cell lines, including 231MFP, A549, HeLa, HEK293T, HEK293A, UM-Chor1, PaCa2, PC3, HUH7, NCI-H460, THP1, SKOV3, U2OS and K562 cells. Some of the chemoproteomic data have been previously reported as part of other studies31,34,48,49,50,51,52,53,54,55,56. All of the isoTOP-ABPP datasets were prepared as previously reported using the IA-alkyne probe15,31,52.
Knockdown studies
RNA interference was performed using siRNA purchased from Dharmacon. CFBE41o-4.7 cells were seeded at 400,000 cells per 6-cm plate and allowed to adhere overnight. Cells were transfected with 33 nM of either non-targeting (ON-TARGETplus Non-targeting Control Pool, Dharmacon, D-001810-10-20) or anti-CFTR siRNA (Dharmacon, L-006425-00-0010) or anti-OTUB1 siRNA (Dharmacon, custom: CCGACUACCUUGUGGUCUAUU) using 8 μl of transfection reagent (DharmaFECT 1 (Dharmacon, T-2001-02), DharmaFECT 4 (Dharmacon, T-2004-02) or Lipofectamine 2000 (Thermo Fisher, 11668027)). Transfection reagent was added to OPTIMEM (Thermo Fisher, 31985070) medium and allowed to incubate for 5 min at room temperature. Meanwhile siRNA was added to an equal amount of OPTIMEM. Solutions of transfection reagent and siRNA in OPTIMEM were then combined and allowed to incubate for 30 min at room temperature. These combined solutions were diluted with complete MEM to provide 33 nM siRNA and 8 μl of transfection reagent per 4 ml of MEM, and the medium was exchanged. Cells were incubated with transfection reagents for 24 h, at which point the medium was replaced with medium containing DMSO or 10 μM NJH-2-057 and incubated for another 24 h. Cells were then collected, and protein abundance was analyzed by western blotting.
Transepithelial conductance assays in human bronchial epithelial cells
Human bronchial epithelial cells from individuals with cystic fibrosis bearing the ΔF508-CFTR mutation were cultured at 37 °C and 5% CO2 in bronchial epithelial cell growth basal medium (BEGM) with SingleQuots Supplements and Growth Factors (Lonza, CC-3170). Cells were maintained in cell culture flasks (Corning, 430641U) for 1 week, and medium was replaced every 2 to 3 d. Cells were washed with Dulbecco’s PBS (Thermo Fisher Scientific, 14040141) and trypsinized for 5 to 10 min with 0.05% Trypsin-EDTA (Thermo Fisher Scientific, 25300120), after which Trypsin Neutralizing Solution (Thermo Fisher Scientific, R002100) was added. Cells were pelleted at 300g for 5 min and resuspended in BEGM with DMEM (Thermo Fisher Scientific, 11965092) and plated at 1×106 cells per plate in 24-well transwell plates (Corning, 3526). Cells were grown submerged in BEGM with DMEM for 1 week with medium changed every 2 to 3 d, at which time they were taken to air liquid interface and grown another 2 weeks before being ready to use.
Cells were treated with either DMSO vehicle, 10 μM VX-809 or 10 μM DUBTAC 24 h before the experiment. Cells were then submerged in Ham’s F12 buffer (Thermo Fisher Scientific, 21700075) with 20 mM HEPES (Thermo Fisher Scientific, 15630080) at pH 7.4 and mounted into the assay system. Transepithelial resistance was recorded using a 24-channel transepithelial current clamp amplifier (TECC24, EP Design). Resistance measurements were taken at intervals of approximately 6 min. Four values were taken to determine baseline resistance, and another four measurements were taken after each of the following additions: 10 μM amiloride (Millipore Sigma, A7410) added apically, 20 µM forskolin (Millipore Sigma, F6886) added apically and 0.5 μM VX770 added both apically and basolaterally. CFTR(inh)-172 (Millipore Sigma, 219672) was then added, and a final six measurements were taken. Transepithelial conductance (G) was calculated from resistance measurements (G = 1/R). Chloride ion transport across the epithelial monolayer is mediated by CFTR, and activation or inhibition of functional CFTR therefore causes changes in transepithelial conductance. In this way, ΔG can be used to measure functional CFTR expression and the functional rescue of CFTR through compound addition.
Quantitative tandem mass tags proteomics analysis
Quantitative TMT-based proteomic analysis was performed as previously described31. Acquired MS data was processed using Proteome Discoverer v.2.4.0.305 software (Thermo) utilizing the Mascot v.2.5.1 search engine (Matrix Science) together with Percolator validation node for peptide–spectral match filtering57. Data were searched against the UniProt protein database (canonical human sequences, EBI) supplemented with sequences of common contaminants. Peptide search tolerances were set to 10 ppm for precursors and 0.8 Da for fragments. Trypsin cleavage specificity (cleavage at lysine, arginine except if followed by proline) allowed for up to two missed cleavages. Carbamidomethylation of cysteine was set as a fixed modification, and methionine oxidation and TMT modification of N termini and lysine residues were set as variable modifications. Data validation of peptide and protein identifications was done at the level of the complete dataset consisting of combined Mascot search results for all individual samples per experiment via the Percolator validation node in Proteome Discoverer. Reporter ion ratio calculations were performed using summed abundances, with the most confident centroid selected from a 20-ppm window. Only peptide-to-spectrum matches that are unique assignments to a given identified protein within the total dataset were considered for protein quantitation. High-confidence protein identifications were reported using a Percolator estimated <1% false discovery rate cutoff. Differential abundance significance was estimated using an analysis of variance test with a Benjamini–Hochberg correction to determine adjusted P values.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated during and/or analyzed during the current study are available as Excel spreadsheets in Supplementary Tables 1–4. Source data are provided with this paper.
Code availability
Data processing and statistical analysis algorithms from our lab can be found on our lab’s GitHub site at https://github.com/NomuraRG.
References
Dixon, S. J. & Stockwell, B. R. Identifying druggable disease-modifying gene products. Curr. Opin. Chem. Biol. 13, 549–555 (2009).
Spradlin, J. N., Zhang, E. & Nomura, D. K. Reimagining druggability using chemoproteomic platforms. Acc. Chem. Res. 54, 1801–1813 (2021).
Nalawansha, D. A. & Crews, C. M. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem. Biol. 27, 998–1014 (2020).
Burslem, G. M. & Crews, C. M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114 (2020).
Schapira, M., Calabrese, M. F., Bullock, A. N. & Crews, C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. 18, 949–963 (2019).
Farnaby, W., Koegl, M., McConnell, D. B. & Ciulli, A. Transforming targeted cancer therapy with PROTACs: a forward-looking perspective. Curr. Opin. Pharmacol. 57, 175–183 (2021).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Takahashi, D. et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell 76, 797–810 (2019).
Yamazoe, S. et al. Heterobifunctional molecules induce dephosphorylation of kinases—a proof of concept study. J. Med. Chem. 63, 2807–2813 (2020).
Siriwardena, S. U. et al. Phosphorylation-inducing chimeric small molecules. J. Am. Chem. Soc. 142, 14052–14057 (2020).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Abbas, T. & Dutta, A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414 (2009).
Li, B. & Dou, Q. P. Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc. Natl Acad. Sci. USA 97, 3850–3855 (2000).
Ward, C. L., Omura, S. & Kopito, R. R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 (1995).
Weerapana, E. et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010).
Backus, K. M. et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 534, 570–574 (2016).
Wiener, R., Zhang, X., Wang, T. & Wolberger, C. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483, 618–622 (2012).
Nakada, S. et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466, 941–946 (2010).
Que, L. T., Morrow, M. E. & Wolberger, C. Comparison of cross-regulation by different OTUB1:E2 complexes. Biochemistry 59, 921–932 (2020).
French, M. E., Koehler, C. F. & Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 7, 6 (2021).
Dunker, A. K. et al. Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 (2001).
Garner, E., Romero, P., Dunker, A. K., Brown, C. & Obradovic, Z. Predicting binding regions within disordered proteins. Genome Inform. Workshop Genome Inform. 10, 41–50 (1999).
Wiener, R. et al. E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat. Struct. Mol. Biol. 20, 1033–1039 (2013).
Riordan, J. R. CFTR function and prospects for therapy. Annu. Rev. Biochem. 77, 701–726 (2008).
Kanner, S. A., Shuja, Z., Choudhury, P., Jain, A. & Colecraft, H. M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 17, 1245–1253 (2020).
Tomati, V. et al. Genetic inhibition of the ubiquitin ligase Rnf5 attenuates phenotypes associated to F508del cystic fibrosis mutation. Sci. Rep. 5, 12138 (2015).
Sondo, E. et al. Pharmacological inhibition of the ubiquitin ligase RNF5 rescues F508del-CFTR in cystic fibrosis airway epithelia. Cell Chem. Biol. 25, 891–905 (2018).
Lopes-Pacheco, M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front. Pharmacol. 10, 1662 (2019).
Rath, A., Glibowicka, M., Nadeau, V. G., Chen, G. & Deber, C. M. Detergent binding explains anomalous SDS–PAGE migration of membrane proteins. Proc. Natl Acad. Sci. USA 106, 1760–1765 (2009).
Yang, Y. et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 11, 433 (2020).
Spradlin, J. N. et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 15, 747–755 (2019).
Zhang, X., Crowley, V. M., Wucherpfennig, T. G., Dix, M. M. & Cravatt, B. F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 15, 737–746 (2019).
Zhang, X. et al. DCAF11 supports targeted protein degradation by electrophilic proteolysis-targeting chimeras. J. Am. Chem. Soc. 143, 5141–5149 (2021).
Ward, C. C. et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol. 14, 2430–2440 (2019).
Ghelli Luserna di Rorà, A., Cerchione, C., Martinelli, G. & Simonetti, G. A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 13, 126 (2020).
Smith, A., Simanski, S., Fallahi, M. & Ayad, N. G. Redundant ubiquitin ligase activities regulate Wee1 degradation and mitotic entry. Cell Cycle 6, 2795–2799 (2007).
Hashimoto, O. et al. Inhibition of proteasome-dependent degradation of Wee1 in G2-arrested Hep3B cells by TGFβ1. Mol. Carcinog. 36, 171–182 (2003).
Li, Z. et al. Development and characterization of a Wee1 kinase degrader. Cell Chem. Biol. 27, 57–65 (2020).
Casement, R., Bond, A., Craigon, C. & Ciulli, A. Mechanistic and structural features of PROTAC ternary complexes. Methods Mol. Biol. 2365, 79–113 (2021).
Gavathiotis, E., Reyna, D. E., Bellairs, J. A., Leshchiner, E. S. & Walensky, L. D. Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 8, 639–645 (2012).
Pryde, D. C. et al. The discovery of potent small molecule activators of human STING. Eur. J. Med. Chem. 209, 112869 (2021).
Ramanjulu, J. M. et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439–443 (2018).
Zorn, J. A. & Wells, J. A. Turning enzymes ON with small molecules. Nat. Chem. Biol. 6, 179–188 (2010).
Chen, L., Liu, S. & Tao, Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct. Target. Ther. 5, 90 (2020).
Riboldi, G. M. & Di Fonzo, A. B. GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8, 364 (2019).
Sarodaya, N., Suresh, B., Kim, K.-S. & Ramakrishna, S. Protein degradation and the pathologic basis of phenylketonuria and hereditary tyrosinemia. Int. J. Mol. Sci. 21, 4996 (2020).
Bartha, I., di Iulio, J., Venter, J. C. & Telenti, A. Human gene essentiality. Nat. Rev. Genet. 19, 51–62 (2018).
Bateman, L. A. et al. Chemoproteomics-enabled covalent ligand screen reveals a cysteine hotspot in reticulon 4 that impairs ER morphology and cancer pathogenicity. Chem. Commun. 53, 7234–7237 (2017).
Chung, C. Y.-S. et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 15, 776–785 (2019).
Boike, L. et al. Discovery of a functional covalent ligand targeting an intrinsically disordered cysteine within MYC. Cell Chem. Biol. 28, 4–13 (2021).
Luo, M. et al. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 28, 559–566 (2021).
Grossman, E. A. et al. Covalent ligand discovery against druggable hotspots targeted by anti-cancer natural products. Cell Chem. Biol. 24, 1368–1376 (2017).
Roberts, A. M. et al. Chemoproteomic screening of covalent ligands reveals UBA5 as a novel pancreatic cancer target. ACS Chem. Biol. 12, 899–904 (2017).
Counihan, J. L., Wiggenhorn, A. L., Anderson, K. E. & Nomura, D. K. Chemoproteomics-enabled covalent ligand screening reveals ALDH3A1 as a lung cancer therapy target. ACS Chem. Biol. 13, 1970–1977 (2018).
Berdan, C. A. et al. Parthenolide covalently targets and inhibits focal adhesion kinase in breast cancer cells. Cell Chem. Biol. 26, 1027–1035 (2019).
Isobe, Y. et al. Manumycin polyketides act as molecular glues between UBR7 and P53. Nat. Chem. Biol. 16, 1189–1198 (2020).
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 4, 923–925 (2007).
Acknowledgements
We thank the members of the Nomura Research Group and Novartis Institutes for BioMedical Research for critical reading of the manuscript. This work was supported by Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for all listed authors. This work was also supported by the Nomura Research Group and the Mark Foundation for Cancer Research ASPIRE Award for D.K.N., N.J.H., L.B., J.N.S., C.C.W. and B.P.B. This work was also supported by grants from the National Institutes of Health (R01CA240981 for D.K.N. and F31CA268278 for N.J.H.) and the National Science Foundation Graduate Fellowship (for L.B.). We also thank H. Celik, A. Lund and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in the CoC-NMR are supported in part by NIH S10OD024998.
Author information
Authors and Affiliations
Contributions
N.J.H., L.B., C.C.W., J.N.S. and D.K.N. conceived of the project idea, provided intellectual contributions, designed experiments, performed experiments, analyzed and interpreted the data and wrote the paper. B.P.B., G.L., E.Z., S.M.B., D.D., R.V.M., L.W.P., D.J.R., F.W., A.O.F., D.F., A.R.E., K.L.R. and A.P. performed experiments, analyzed and interpreted data and provided intellectual contributions. M.H., L.M.M., J.M.M., M.S. and J.A.T. provided intellectual contributions to the project and overall design of the project. Experimentally, C.C.W. and J.N.S. performed covalent ligand screens to identify EN523 and performed characterization of EN523 interactions with OTUB1. N.J.H. and L.B. characterized EN523 interactions with OTUB1, synthesized the DUBTACs and tested, characterized and validated the DUBTACs in cells. K.L.R. and A.P. assisted in DUBTAC synthesis and testing in cells. A.R.E. tested DUBTACs in cells. D.K.N. performed MS experiments, designed experiments and analyzed and interpreted the data. B.P.B. performed proteasome inhibitor experiments. G.L. and J.M.M. designed and synthesized DUBTACs with rigid linkers. E.Z. synthesized EN523 analogs. S.M.B. performed quantitative proteomics experiments. D.D., A.O.F. and F.W. performed biochemical and NMR studies on EN523 interactions with OTUB1. L.M.M. and D.F. performed native MS experiments. R.V.M., L.W.P. and D.J.R. performed CFTR transepithelial conductance studies. M.S., J.A.T. and D.K.N. conceived experiments and supervised the work.
Corresponding author
Ethics declarations
Competing interests
J.A.T., J.M.M., L.M.M., D.D., M.H., M.S., S.M.B., G.L., R.V.M., L.W.P., D.J.R., F.W., A.O.F. and D.F. are employees of Novartis Institutes for BioMedical Research. This study was funded by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. D.K.N. is a cofounder, shareholder and adviser for Frontier Medicines and Vicinitas Therapeutics. D.K.N. is also on the scientific advisory board of The Mark Foundation for Cancer Research and Photys Therapeutics.
Peer review
Peer review information
Nature Chemical Biology thanks Alessio Ciulli, Doug Johnson and Feng Wang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Primary covalent ligand screen against OTUB1.
(a) Analysis of aggregate chemoproteomic data for DUBs. Top 10 candidate DUBs described in Fig. 1c for total aggregate spectral counts of the particular probe-modified cysteine found in our aggregate chemoproteomic data showing OTUB1 C23 appears far more frequently in chemoproteomic datasets compared to the other DUBs. (b) C23 belongs to an intrinsically disordered region within OTUB1 as assessed by PONDR. (c) Covalent ligand screen of cysteine-reactive libraries competed against IA-rhodamine labeling of recombinant OTUB1 to identify binders to OTUB1 by gel-based ABPP. Vehicle DMSO or cysteine-reactive covalent ligands (50 μM) were pre-incubated with OTUB1 for 30 min at room temperature prior to IA-rhodamine labeling (500 nM, 30 min room temperature). OTUB1 was then separated by SDS/PAGE and in-gel fluorescence was assessed and quantified. This screen was performed with n = 1 biologically independent samples/group. Any hits showing >50 % loss of IA-rhodamine labeling were subsequently re-tested for reproducibility. EN523 was the only reproducible hit from this primary screen. Raw gels can be found in Source data.
Extended Data Fig. 2 NMR analysis of OTUB1, EN523, and UBE2D2.
(a) 13C-HMQC spectrum of OTUB1 labeled on methyl groups of isoleucine, alanine, valine and leucine residues. The presence of peaks with negative proton chemical shifts indicates that the protein is properly folded. (b) Overlay of HMQC spectra of apo-OTUB1 (black) and EN523-bound OTUB1 (red). While both spectra are mostly identical, we identified small but clear chemical shift perturbations of alanine, isoleucine, valine and leucine peaks. Some of these signal changes are shown in the respective blow-up boxes. (c) Overlay of HMQC spectra of apo-OTUB1 (black), UBE2D2 bound OTUB1 (red) and EN523/UBE2D2-bound OTUB1 (blue). The strong chemical shift perturbations (CSPs) are evidence of specific interactions between OTUB1 and the ubiquitinylated ubiquitin-conjugating enzyme. The lack of significant differences between spectra recorded in the presence and absence of EN523 prove that the covalent ligand does not interfere with the protein-protein interaction. Differing peak shift pattern are only seen for peaks directly affected by compound binding (see inlay for blow-up of Ala region). (d) Overlay of HMQC spectra of apo-OTUB1 (black), Ub-UBE2D2 bound OTUB1 (red) and EN523/Ub-UBE2D2-bound OTUB1 (blue). The strong CSPs are evidence of specific interactions between OTUB1 and the ubiquitin-conjugating enzyme. The lack of significant differences between spectra recorded in the presence and absence of EN523 prove that the covalent ligand does not interfere with the protein-protein interaction. Differing peak shift pattern are only seen for peaks directly affected by compound binding (see inlay for blow-up of Ala region).
Extended Data Fig. 3 Structure-activity relationships of EN523 analogs with OTUB1.
Gel-based ABPP analysis EN523 analogs against OTUB1. Vehicle DMSO or EN523 analogs were pre-incubated with recombinant OTUB1 for 30 min at 37 °C prior to IA-rhodamine labeling (100 nM, 30 min room temperature). OTUB1 was then separated by SDS/PAGE and in-gel fluorescence was assessed. Also shown is silver staining showing protein loading. Shown are representative gels of n = 3 biologically independent samples/group. Raw gels can be found in Source data.
Extended Data Fig. 4 EN523 does not alter OTUB1 levels and NJH-2-075 engages OTUB1 in CFBE41o-4.7 cells expressing ΔF508-CFTR.
(a) CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or EN523 (10 μM) for 24 h and OTUB1 and loading control vinculin levels were assessed by Western blotting. (b) NJH-2-075 engagement of OTUB1 in CFBE41o-4.7 cells expressing ΔF508-CFTR. Cells were treated with DMSO vehicle or NJH-2-075 (50 μM) for 2 h, after which cell lysates were subjected to CuAAC with biotin picolyl azide and NJH-2-075 labeled proteins were subjected to avidin pulldown, eluted, separated by SDS/PAGE, and blotted for OTUB1 and vinculin. Both input lysate and pulldown levels are shown. Blots shown are representative blots from n = 3 biologically independent samples/group. Raw blots can be found in Source data.
Extended Data Fig. 5 Effect of DUBTACs on mutant CFTR levels.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 and CFTR and loading control GAPDH levels were assessed by Western blotting. For dose-response studies, NJH-2-057 was treated for 24 h. For time-course studies, NJH-2-057 was treated at 10 μM. Dose-response and time-course data gels are representative of n = 3 biologically independent samples/group and are quantified in the bar graphs to the right. Data in bar graphs show individual biological replicate values and average ± sem from n = 3 biologically independent samples/group. Raw blots and bar graph data can be found in Source data.
Extended Data Fig. 6 Effect of DUBTACs on mutant CFTR levels.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 and CFTR and loading control GAPDH levels were assessed by Western blotting using three different antibodies against CFTR from the ones used for the main figures. NJH-2-057 was treated for 24 h. Gels are representative of n = 3 biologically independent samples/group. Raw blots can be found in Source data.
Extended Data Fig. 7 Effect of DUBTACs on mutant CFTR levels in siControl and siCFTR cells.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 (10 μM) for 24 h and CFTR and loading control GAPDH levels were assessed by Western blotting. Blot is representative of n = 3 biologically independent samples/group. Raw blots can be found in Source data.
Extended Data Fig. 8 Linker dependence of CFTR DUBTACs.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or DUBTACs (10 μM) for 24 h and CFTR and loading control GAPDH levels were assessed by Western blotting. Blot is representative of n = 3 biologically independent samples/group. Raw blots can be found in Source data.
Extended Data Fig. 9 CFTR DUBTACs with rigid linkers.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or DUBTACs (10 μM) for 24 h and CFTR and loading control actin levels were assessed by Western blotting. Blot is representative of n = 3 biologically independent samples/group. Bar graphs show quantification of CFTR levels shown as individual biological replicate data and average ± sem with n = 3 biologically independent samples/group. Statistical significance was calculated with unpaired two-tailed Student’s t-tests compared to vehicle-treated controls and is expressed as *p < 0.05. Raw blots, bar graph, and exact p-values can be found in Source data.
Extended Data Fig. 10 Negative control DUBTACs.
CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or compounds (10 μM) for 24 h and CFTR and loading control GAPDH levels were assessed by Western blotting. Blot is representative of n = 3 biologically independent samples/group. Raw blots can be found in Source data.
Supplementary information
Supplementary information
Supplementary Figs. 1–4 and Note.
Supplementary Table 1
Chemoproteomic analysis of DUBs. Table of all probe-modified cysteines identified for 65 DUBs from 455 distinct isoTOP-ABPP experiments. These data are from our research group’s aggregate chemoproteomic experiments from various human cell lines wherein cell lysates were labeled with an IA-alkyne probe and taken through the isoTOP-ABPP procedure. Shown are the DUBs, probe-modified peptides, the site of the modified cysteine, the aggregate spectral counts for each particular probe-modified site identified across 455 experiments and the total numbers of experiments for which the probe-modified peptide was observed.
Supplementary Table 2
Structures of covalent ligands screened against OTUB1. Covalent ligand screen of cysteine-reactive libraries competed against IA-rhodamine labeling of recombinant OTUB1 to identify binders to OTUB1 by gel-based ABPP. Vehicle DMSO or cysteine-reactive covalent ligands (50 μM) were preincubated with OTUB1 for 30 min at room temperature before IA-rhodamine labeling (500 nM for 30 min at room temperature). OTUB1 was then separated by SDS–PAGE, and in-gel fluorescence was assessed and quantified. Gel-based ABPP data are shown in Extended Data Fig. 1a. Quantification of gel-based ABPP data normalized to the average of DMSO vehicle-treated controls in each gel is shown in tab 1.
Supplementary Table 3
TMT-based quantitative proteomic profiling of NJH-2-057 treatment. CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 (tabs 1 and 2) or EN523 or lumacaftor (tabs 3 and 4) (10 μM) for 24 h. Data shown are from n = 3 biologically independent samples per group.
Supplementary Table 4
IsoTOP-ABPP analysis of NJH-2-057. CFBE41o-4.7 cells expressing ΔF508-CFTR were treated with vehicle DMSO or NJH-2-057 (10 μM) for 8 h. Resulting cell lysates were labeled with IA-alkyne (200 μM) for 1 h and taken through the isoTOP-ABPP procedure Shown in red are the probe-modified peptides that showed isotopically light/heavy or control/NJH-2-57 ratios >4 with adjusted P values of <0.05. The data are from n = 3 biologically independent samples per group.
Source data
Source Data Fig. 2
Unprocessed western blots and gels.
Source Data Fig. 3
Unprocessed western blots and gels.
Source Data Fig. 3
Raw and statistical data.
Source Data Fig. 4
Unprocessed western blots and gels.
Source Data Fig. 4
Raw and statistical data.
Source Data Fig. 5
Unprocessed western blots and gels.
Source Data Fig. 5
Raw and statistical data.
Source Data Extended Data Fig. 1
Unprocessed gels.
Source Data Extended Data Fig. 3
Unprocessed gels.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 5
Raw data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Unprocessed western blots.
Source Data Extended Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 9
Unprocessed western blots.
Source Data Extended Data Fig. 9
Raw data.
Source Data Extended Data Fig. 10
Unprocessed western blots.
Rights and permissions
About this article

Cite this article
Henning, N.J., Boike, L., Spradlin, J.N. et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat Chem Biol 18, 412–421 (2022). https://doi.org/10.1038/s41589-022-00971-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-022-00971-2
This article is cited by
-
Proteome-scale discovery of protein degradation and stabilization effectors
Nature (2024)
-
DNA-encoded library-enabled discovery of proximity-inducing small molecules
Nature Chemical Biology (2024)
-
Emerging roles of deubiquitinating enzymes in actin cytoskeleton and tumor metastasis
Cellular Oncology (2024)
-
OTUB1 accelerates hepatocellular carcinoma by stabilizing RACK1 via its non-canonical ubiquitination
Cellular Oncology (2024)
-
Linking ATP and allosteric sites to achieve superadditive binding with bivalent EGFR kinase inhibitors
Communications Chemistry (2024)
