
Abstract
In December 2019, the first cases of infection with a novel coronavirus, SARS-CoV-2, were diagnosed. Currently, there is no effective antiviral treatment for COVID-19. To address this emerging problem, we focused on the SARS-CoV-2 main protease that constitutes one of the most attractive antiviral drug targets. We have synthesized a combinatorial library of fluorogenic substrates with glutamine in the P1 position. We used it to determine the substrate preferences of the SARS-CoV and SARS-CoV-2 main proteases. On the basis of these findings, we designed and synthesized a potent SARS-CoV-2 inhibitor (Ac-Abu-dTyr-Leu-Gln-VS, half-maximal effective concentration of 3.7 µM) and two activity-based probes, for one of which we determined the crystal structure of its complex with the SARS-CoV-2 Mpro. We visualized active SARS-CoV-2 Mpro in nasopharyngeal epithelial cells of patients suffering from COVID-19 infection. The results of our work provide a structural framework for the design of inhibitors as antiviral agents and/or diagnostic tests.

Similar content being viewed by others

Main
In December 2019, a severe respiratory disease of unknown origin emerged in Wuhan, Hubei province, China1,2. Symptoms of the first patients were flu-like and included fever, cough and myalgia, but with a tendency to develop a potentially fatal dyspnea and acute respiratory distress syndrome2. Genetic analysis confirmed a betacoronavirus as the causing agent. The virus was initially named 2019 novel coronavirus (2019-nCoV)1,2,3, but shortly thereafter, it was renamed SARS-CoV-2 (ref. 4). By 28 August 2020, the World Health Organization had registered >24 million cumulative cases of coronavirus disease 2019 (COVID-19), with 836,475 deaths5.
Currently, there is no approved vaccine or treatment for COVID-19. Efforts are being made to characterize molecular targets, pivotal for the development of anticoronaviral therapies6. The main protease (Mpro, also known as 3CLpro), is one of the coronavirus nonstructural proteins (Nsp5) designated as a potential target for drug development7,8. Mpro cleaves the viral polyproteins, generating 12 nonstructural proteins (Nsp4-Nsp16), including the RNA-dependent RNA polymerase (RdRp, Nsp12) and the helicase (Nsp13). Inhibition of Mpro would prevent the virus from replication and therefore constitutes one of the potential anticoronaviral strategies7,8,9.
Due to the close phylogenetic relationship between SARS-CoV-2 and SARS-CoV3,10,11, their main proteases share many structural and functional features. From the perspective of the design and synthesis of new Mpro inhibitors, a key feature of both the enzymes is their ability to cleave the peptide bond following Gln. The SARS-CoV Mpro cleaves polyproteins mainly within the Leu-Gln↓(Ser, Ala, Gly) sequence (↓ indicates the cleavage site), which appears to be a preferred pattern of this protease7,9,12. The specificity for peptide bond hydrolysis after Gln residues is also observed for main proteases of other coronaviruses13,14 but is rare for human enzymes. This observation, along with further studies on the Mpro, can potentially lead to new broad-spectrum anticoronaviral inhibitors with minimal side effects15.
In the present study, we applied the HyCoSuL (hybrid combinatorial substrate library) approach to determine the full substrate specificity profile of SARS-CoV and SARS-CoV-2 Mpros. The use of natural and a large number of unnatural amino acids with diverse chemical structures allowed an in-depth characterization of the residue preference of the binding pockets within the substrate-binding site of the proteases. The results from library screening enabled us to design and synthesize ACC-labeled substrates with improved catalytic efficiency in comparison to a substrate containing only natural amino acids. Moreover, results from our studies clearly indicate that SARS-CoV Mpro and SARS-CoV-2 Mpro exhibit highly overlapping substrate specificities. We have used this knowledge to design activity-based probes (ABPs) specific for the SARS-CoV main proteases as well as tetrapeptide inhibitors. Further, we present a crystal structure of the SARS-CoV-2 Mpro in complex with one of the ABPs. Finally, using the ABP, we were able to visualize active SARS-CoV-2 Mpro within nasopharyngeal epithelial cells of patients with active COVID-19 infection. These data provide a useful basis for the design of chemical compounds for effective diagnosis and therapy of COVID-19.
Results
Substrate specificity of SARS-CoV and SARS-CoV-2 Mpros
To determine the SARS-CoV Mpro and SARS-CoV-2 Mpro substrate preferences, we applied a HyCoSuL approach. The library consists of three sublibraries, each of them comprising a fluorescent tag, ACC (7-amino-4-carbamoylmethylcoumarin); two fixed positions and two varied positions containing an equimolar mixture of 19 amino acids (Mix) (P2 sublibrary, Ac-Mix-Mix-X-Gln-ACC; P3 sublibrary, Ac-Mix-X-Mix-Gln-ACC; P4 sublibrary, Ac-X-Mix-Mix-Gln-ACC, X = 19 natural and over 100 unnatural amino acids, Extended Data Fig. 1). We incorporated glutamine at the P1 position, because the available crystal structures of SARS-CoV Mpro revealed that only glutamine (and, at only one cleavage site, histidine) can occupy the S1 pocket of this enzyme12,16. The imidazole of His163, located near the bottom of the S1 pocket, is suitably positioned to interact with the Gln side chain. The Gln is also involved in two other interactions; that is, with the main chain of Phe140 and the side chain of Glu166. The library screen revealed that SARS-CoV and SARS-CoV-2 Mpro display very similar substrate specificities (Fig. 1). The most preferred amino acid at the P2 position is leucine in the case of both proteases. SARS-CoV Mpro exhibits lower activity toward other tested amino acids at this position (<30%). Leu selectivity for SARS-CoV Mpro is in high agreement with a previous report by Zhu et al.17. However, we have noticed some discrepancies in the level of selectivity for other natural amino acids. This is the result of differences in the substrate specificity profiling approach, where we are using a combinatorial mixture of natural amino acids, while Zhu et al. were using a defined peptide sequence. Nevertheless, in both approaches, preferences for the same natural amino acids were observed. The S2 pocket of both investigated enzymes can accommodate other hydrophobic residues, such as 2-Abz, Phe(4-NO2), 3-Abz, β-Ala, Dht, hLeu, Met and Ile (amino-acid structures are presented in Supplementary Table 1). At the P3 position, both enzymes prefer hydrophobic d and l amino acids and also positively charged residues; the best are Tle, d-Phe, d-Tyr, Orn, hArg, Dab, Dht, Lys, d-Phg, d-Trp, Arg and Met(O)2. At the P4 position, SARS-CoV and SARS-CoV-2 Mpro possess broad substrate specificity. The most preferred are small aliphatic residues such as Abu, Val, Ala and Tle, but other hydrophobic amino acids are also accepted. These findings can be partly explained by the available crystal structures of SARS-CoV Mpro in complex with inhibitors8,12,16. The hydrophobic S2 subsite of SARS-CoV Mpro is more flexible compared to alphacoronavirus Mpros, which explains the less stringent specificity15. The S2 pocket can form hydrophobic interactions with P2 residues that are not only limited to leucine. The S3 pocket of SARS-CoV Mpro is not well defined, which is also reflected by our P3 substrate specificity profile. The S4 pocket can be occupied by small residues due to the crowded cavity formed by Leu167, Pro168 at the bottom and Thr190, Ala191 at the top wall.

The average relative activity is presented as a percentage of the best-recognized amino acid (n = 2, where n represents the number of independent experiments).
To validate the results from library screening, we designed and synthesized ACC-labeled substrates containing the most preferred amino acids in each position. Then, we measured the rate of substrate hydrolysis (chemical compounds 1–8) relevant to each protease (Extended Data Fig. 2). The data clearly demonstrate that SARS-CoV Mpro and SARS-CoV-2 Mpro exhibit the same activity toward tested substrates. The results are consistent with the HyCoSuL screening data. The most preferred substrate, Ac-Abu-Tle-Leu-Gln-ACC (1), is composed of the best amino acids in each position (Supplementary Table 2). Kinetic parameters were determined for the two best substrates (Ac-Abu-Tle-Leu-Gln-ACC, 1; Ac-Thz-Tle-Leu-Gln-ACC, 2) and one containing the best-recognized natural amino acids (Ac-Val-Lys-Leu-Gln-ACC, 3) (Table 1 and Extended Data Fig. 3) toward SARS-CoV-2 Mpro. Due to substrate precipitation because of high concentration needed in the assay, kinetic parameters toward SARS-CoV Mpro could not be determined. Analysis of kinetic parameters revealed that these three substrates differ in the kcat value, while KM values are comparable. Since SARS-CoV-2 Mpro exhibits low activity toward tetrapeptide substrates, we synthesized internally quenched fluorescent substrates (IQF) containing the same amino acids at the P4-P1 positions as substrates QS1, QS2 and QS4. The general sequence of IQF substrates was derived from the N-terminal autocleavage sequence of the viral protease reported previously (MCA-AVLQSGFRK(Dnp)K)18; instead of the MCA fluorophore, ACC was used. We also incorporated a small linker (glycine residue) between ACC and the amino acid at the P4 position for efficient synthesis. Specificity constant values (kcat/KM) for these IQF substrates were much higher than for tetrapeptide substrates. IQF substrates containing the same amino acids at P4-P1 positions as substrates QS1 (ACC-Gly-Abu-Tle-Leu-Gln-Ser-Gly-Phe-Arg-Lys(dnp)-Lys-NH2, 9) and QS4 (ACC-Gly-Thz-Tle-Leu-Gln-Ser-Gly-Phe-Arg-Lys(dnp)-Lys-NH2, 10) were better recognized than the IQF substrate possessing the same peptide sequence as the QS2 (ACC-Gly-Val-Lys-Leu-Gln-Ser-Gly-Phe-Arg-Lys(dnp)-Lys-NH2, 11) substrate (Supplementary Table 3). This is in line with kinetic results obtained for tetrapeptide substrates.
Inhibitors and ABPs of SARS-CoV-2 Mpro
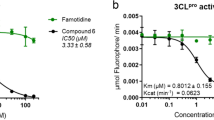
In the next step, the best substrate QS1 was converted into an inhibitor and ABPs. The inhibitor contained the acetylated peptide sequence and vinyl sulfone as an irreversible reactive group (Ac-Abu-Tle-Leu-Gln-VS, Ac-QS1-VS (12), Fig. 2a). The probes included an N-terminal biotin tag (B) or the Cyanine 5 dye (Cy5), polyethylene glycol (PEG(4)) as a linker, the best peptide sequence and vinyl sulfone (Fig. 2a, and Extended Data Fig. 4a). To evaluate the sensitivity of the designed probes, we performed SDS–PAGE analysis followed by protein transfer onto membranes and ABP visualization. We observed SARS-CoV-2 Mpro (100 nM) labeling by B-QS1-VS (13) at a concentration of 200 nM and by Cy5-QS1-VS (14) at 100 nM (Fig. 2b and Extended Data Fig. 4b) that reflected the results of the kobs/I (I, inhibitor or probe concentration) analysis (Table 2 and Extended Data Fig. 5). In our previous studies, we showed that an ABP with low potency can be applied to detect protease activity in cell assays19. To determine the probe selectivity, we performed cell lysate assays. A HeLa lysate was incubated with different probe concentrations (50, 100 and 200 nM) (Extended Data Fig. 4c). The cell lysate experiment confirmed probe selectivity. To verify that unknown bands (about 30 kDa and between 49 and 62 kDa) were due to unspecific Cy5 labeling, we incubated the cell lysate with inhibitor (Ac-QS1-VS) for 30 min at 37 °C and then with Cy5-QS1-VS for 15 min at 37 °C (lanes 8–10 on the membrane, Extended Data Fig. 4c). The same protein bands were observed on the membrane when cell lysates were incubated with and without Ac-QS1-VS, which confirmed unspecific protein labeling by the Cy5 dye. Due to this unspecific protein labeling by Cy5-QS1-VS, we decided to slightly modify the probe structure. Since amino acids with d-stereochemistry are not recognized by many human proteases20,21,22,23, we changed Tle at the P3 position to d-Tyr, which is well recognized by SARS-CoV-2 Mpro at this position, to improve probe selectivity. The Cy5 fluorophore was exchanged to the Bodipy FL fluorophore. The designed inhibitor (Ac-Abu-dTyr-Leu-Gln-VS, Ac-QS5-VS, 15) and the ABP (Bodipy-PEG(4)-Abu-dTyr-Leu-Gln-VS, Bodipy-QS5-VS, 16) were tested on the recombinant enzyme and cell lysates (Fig. 2). Kinetic analysis revealed that Ac-QS5-VS exhibited the same potency toward SARS-CoV-2 Mpro as Ac-QS1-VS (Table 2). The designed ABP, Bodipy-QS5-VS, was slightly less active toward the main protease than Cy5-QS1-VS. To assess Bodipy-QS5-VS sensitivity, we incubated SARS-CoV-2 Mpro (at three different concentrations of 5, 10 and 50 nM) with different probe concentrations for 30 min. We observed the detection of SARS-CoV-2 Mpro at 5 nM by 2.5 µM Bodipy-QS5-VS (Fig. 2c). The enzyme labeling by the ABP is noticeable after 5 min of incubation (Fig. 2d). Cell lysate assays (HeLa and human embyronic kidney (HEK)-293T) confirmed Bodipy-QS5-VS selectivity (Fig. 2e). Ac-Abu-dTyr-Leu-Gln-VS was also tested for its antiviral activity using a fully replication-competent SARS-CoV-2 strain in Huh7 cell culture. Here the inhibitor showed selective inhibition (half-maximal effective concentration (EC50) = 3.7 µM; interquartile range (IQR) 3.5–4.0 µM) with no notable toxicity (50% cytotoxic concentration (CC50) > 100 µM). As a comparison, the parent nucleoside of remdesivir (GS-441524) under the same conditions showed an EC50 of 1.15 µM (IQR 1.1–1.2 µM) and a CC50 of 87 µM (IQR 82–92 µM) (Fig. 2f,g).

a, Structure of inhibitors and ABPs. b–d, SARS-CoV-2 Mpro labeling by probes B-QS1-VS (b) and Bodipy-QS5-VS) (c). c, SARS-CoV-2 Mpro (5, 10, and 50 nM) was detected by different concentrations of Bodipy-QS5-VS. d, Time-point analysis of Bodipy-QS5-VS binding to the enzyme. e, SARS-CoV-2 Mpro probe selectivity in HeLa and HEK-293T lysates (asterisk shows the SARS-CoV-2 Mpro, which was spiked into the cell lysates). The cell lysate was incubated with or without the inhibitor Ac-QS5-VS for 30 min at 37 °C; next, different probe concentrations were added and the samples were incubated for 30 min at 37 °C. The biotinylated ABP was detected with a fluorescent streptavidin Alexa Fluor 647 conjugate (1:10,000) in TBS-T with 1% BSA using an Azure Biosystems Sapphire Biomolecular Imager (laser 658 and 488 nm for Bodipy FL dye) and the Azure Spot Analysis software. Each experiment was repeated independently two times with similar results. f,g, Antiviral activity on SARS-CoV-2 (f) and cellular toxicity (g) in Huh7 cells of Ac-Abu-dTyr-Leu-Gln-VS and GS-441524. Antiviral activity and toxicity was determined by quantification of viability of infected (f) or uninfected (g) cell cultures (see Methods). Two independent dose–response curves were investigated for each compound (indicated by the circles and triangels). Nonlinear fitting of the dose–response curves results in EC50s of 1.2 and 1.3 µM for GS-441524 and 3.5 and 4.0 µM for Ac-Abu-dTyr-Leu-Gln-VS. Calculated CC50s are 69 and 95 µM for GS-441524 and >100 µM for Ac-Abu-dTyr-Leu-Gln-VS.
Crystal structure of SARS-CoV-2 Mpro with B-QS1-VS
To visualize the steric details of the interactions between the Mpro and the ABP, Biotin-PEG(4)-Abu-Tle-Leu-Gln-VS (B-QS1-VS), we determined the X-ray crystal structure of the complex between the two components. The probe was cocrystallized with the recombinant and highly purified SARS-CoV-2 Mpro. Crystals diffracted to 1.7 Å resolution and were of space group P6122, with one ABP-Mpro monomer per asymmetric unit (see Supplementary Table 4 for crystallographic details). The structure was refined to reasonable R factors and good geometry. Most of the atoms of the ABP were clearly seen in the 2Fo–Fc electron density (Fo, observed structure factor amplitudes; Fc, structure factor amplitudes calculated from the model) at a contour level of 0.5σ. The reason for this is that the flexible tail of the ABP, the PEG(4) chain and the terminal biotin label, are in contact with a neighboring Mpro dimer in the crystal lattice (Extended Data Fig. 6a). This is of course of little relevance for the situation in solution; hence we discuss only the interaction of the P4-P1 residues with the parent Mpro molecule (Fig. 3) here. The oxygen atoms of the vinyl sulfone group point toward the oxyanion hole of the protease and accept hydrogen bonds (2.81 and 3.26 Å) from the main-chain amide groups of Gly143 and Cys145, respectively. Also, the side-chain amide nitrogen of Asn142 donates a 2.90-Å hydrogen bond (H bond) to one of these oxygens. The methyl group attached to the sulfone makes hydrophobic contacts with the Cγ2 atom of Thr25 and the side chain of Leu27, within the S1' subsite. The catalytic cysteine residue is covalently linked to the Cβ atom of the vinyl group, at a distance of 1.83 Å.

The Fo–Fc electron density for residues P4-P1’ of the ABP is shown at a contour level of 2.5σ. All parts of the ABP beyond the P4 residue would be outside the parent Mpro dimer and have been omitted from this figure. However, the rest of the ABP interacts with a neighboring Mpro dimer in the crystal (Extended Data Fig. 6a) and is seen in the electron density maps.
The P1-glutamine side chain makes the expected interaction with His163 (2.72 Å, through the Oε1 atom) and the Phe140 main-chain oxygen (3.22 Å, through Nε2) and Glu166 Oε1 (3.25 Å). The Nε2 of the P1-Gln thus donates a three-center (bifurcated) hydrogen bond to these two acceptors. This is also reflected in inhibitors that carry a γ-lactam as Gln surrogate in the P1 position, such as compound 13b (ref. 9). The Leu residue in the S2 pocket makes the canonical interactions previously observed17, with residues Met49, Met165, His41 and the Cα atom of Arg188. Also, the main-chain amide of the P2-Leu donates a 2.98-Å H bond to the side chain Oε1 of Gln189.
There is no well-defined pocket for the P3 moiety, which is therefore mostly solvent-exposed. There may be some weak interaction of the Tle side chain with the hydrophobic portion of the neighboring Glu166 side chain. The polar main-chain atoms of the P3 residue form hydrogen bonds with the protein main chain at Glu166. The aminobutyric acid in P4 makes weak hydrophobic interactions with Leu167 and Gln189, and its main-chain NH group donates a H bond to Thr190 O.
We have previously noticed that compared to the SARS-CoV Mpro, Thr285 has been replaced by Ala in SARS-CoV-2 Mpro and the neighboring Ile286 by Leu9. In the SARS-CoV Mpro, the Thr285 makes a hydrogen bond with its symmetry-mate across the twofold axis creating the Mpro dimer. The loss of this H bond in the SARS-CoV-2 Mpro enables the monomers of the dimer to approach each other more closely. In the crystal structure presented here, the space between the two protomers generated by the mutation is filled by a chloride ion adopted from the crystallization buffer (Extended Data Fig. 6b). This might be taken as a hint for this region around residues 284–286 being a hotspot for mutations, due to nonideal packing of the two protomers of the dimer at this point.
SARS-CoV-2 Mpro detection and imaging in patient samples
Fluorescent-tagged ABPs currently represent the classic standard in terms of application for labeling of biological samples and have been successfully used for visualization of many proteases in the past24,25,26. We wanted to see if the Cy5-QS1-VS developed by us can be used for detection of SARS-CoV-2 Mpro in human samples. Thus, we recruited one patient with mild symptoms of COVID-19, who was positive for SARS-CoV-2 RNA in two independent quantitative PCR with reverse transcription assays validated for diagnostic purposes. We incubated the probe at final concentration of 1 μM with cells collected from nasopharyngeal swabs of the patient on the first and fifth days after diagnosis and subjected them to confocal laser scanning microscopy. In parallel, we carried out the same experiment with a healthy donor (COVID-19-negative control). These serial measurements revealed that 10–15% of the cells were positive for staining with the Cy5-QS1-VS probe in the patient who tested positive for COVID-19 (Extended Data Fig. 7). A particularly strong signal from SARS-CoV-2 Mpro was observed on the fifth day after diagnosis. No signal from SARS-CoV-2 Mpro labeling was observed in the sample from the healthy donor (Extended Data Fig. 7). Due to a noticeable background fluorescence signal from the Cy5 dye in the control sample (negative for COVID-19), we changed the probe structure and performed the same experiments using a new probe, Bodipy-QS5-VS, among additional six patients positive for SARS-CoV-2 RNA with clinical signs of COVID-19 and three negative, healthy individuals (control samples). We observed labeling of SARS-CoV-2 Mpro in the patients positive for COVID-19 by Bodipy-QS5-VS and no signal from the probe in the patients who tested negative (Fig. 4 and Extended Data Fig. 8a,c). When the cells (samples positive for COVID-19) were incubated with Ac-QS1-VS inhibitor before the Bodipy-QS5-VS probe, SARS-CoV-2 Mpro staining was reduced (Extended Data Fig. 8b). Thus, we were able to show that human cells collected ex vivo contain active SARS-CoV-2 Mpro during SARS-CoV-2 infection.

Confocal microscopy of the epithelial cells of nasopharyngeal swabs co-stained with Bodipy-QS5-VS SARS-CoV-2 Mpro probe and anti-ACE2 antibody (ACE2, angiotensin-converting enzyme 2) with Cy5 AffiniPure donkey anti-Goat secondary antibody and DAPI. Due to ethical concerns, one nasopharyngeal swab was collected from each patient. Cells come from the following patients who tested positive: P01DZ, P02JB and P03KB and a control (CTRL). Scale bars, 25 µm. Control 1; scale bars, 50 µm.
Discussion
SARS-CoV-2, first observed in Wuhan, China, caused the current pandemic of COVID-19 disease. The lack of a vaccine or approved medications for direct treatment of the disease has led to strenuous efforts to find therapies and stop the pandemic. Among the promising therapeutic targets are two viral proteases: SARS-CoV-2 Mpro and SARS-CoV-2 PLpro. The first, SARS-CoV-2 Mpro, is used by the virus for protein maturation and its structure has been recently described9. Moreover, the results of retargeting about 10,000 drugs, drug candidates in clinical trials and other bioactive compounds resulted in selection of several candidates as potential inhibitors of this enzyme18. In our research, we decided to thoroughly examine SARS-CoV-2 Mpro to find the optimal chemical tools in the form of substrates, inhibitors and ABPs. First, we obtained a targeted library of fluorogenic substrates (HyCoSuL) toward this protease and determined the substrate specificity at the P4-P2 positions. We directly compared the substrate specificity of the main protease with the same protease from the previous SARS coronavirus, which caused an epidemic in 2003. Our data clearly demonstrate that these enzymes have very similar preferences for natural and unnatural amino acids at the P4-P2 positions. They tolerate many different amino acids at P4 and P3, and have a strong preference for Leu at P2. This information is certainly crucial for the aspect of drug retargeting, but so is the use of information obtained in previous years for SARS-CoV Mpro to be used for current research. In the next step, we created potent inhibitors and ABPs with sequences based on the HyCoSuL screening results. This strategy had been successfully applied in our previous studies to design selective inhibitors and ABPs for cysteine24,27, serine22 and threonine23 proteases. In the case of B-QS1-VS, we also obtained a crystal structure of its complex with SARS-CoV-2 Mpro, which accurately shows the binding mechanism in the P4-P1’ pockets. This knowledge certainly complements the information already obtained for other inhibitor molecules published for this enzyme. In turn, we used the fluorescent ABP to visualize the SARS-CoV-2 Mpro activity in patient samples with COVID-19, thus confirming that it is an excellent tool that can be used to detect this enzyme and can also be used as a diagnostic tool.
In conclusion, our research allowed for the exact characterization of SARS-CoV-2 Mpro in terms of both aminoacid preferences as well as the design of targeted inhibitors and ABPs. The reagents described here can be used to optimize structures that lead to anti-COVID-19 drugs, as well as for further drug retargeting.
Methods
Reagents
The reagents used for solid-phase peptide synthesis were as follows: Rink amide resin (particle size 100–200 mesh, loading 0.74 mmol g−1), all Fmoc-amino acids, O-benzotriazole-N,N,N’,N’-tetramethyluronium-hexafluorophosphate (HBTU), 2-(1-H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU), piperidine, diisopropylcarbodiimide (DICI) and trifluoroacetic acid (TFA), purchased from Iris Biotech; anhydrous N-hydroxybenzotriazole (HOBt) from Creosauls; 2,4,6-collidine (2,4,6-trimethylpyridine), high-performance liquid chromatography- (HPLC-)grade acetonitrile, triisopropylsilane (TIPS) from Sigma-Aldrich and N,N-diisopropylethylamie (DIPEA) from VWR International. N,N-dimethylformamide (DMF), dichloromethane (DCM), methanol (MeOH), diethyl ether (Et2O), acetic acid (AcOH) and phosphorus pentoxide (P2O5), obtained from Avantor. Designed substrates were purified by HPLC on a Waters M600 solvent delivery module with a Waters M2489 detector system using a semipreparative Wide Pore C8 Discovery column. The solvent composition was as follows: phase A (water:0.1% TFA) and phase B (acetonitrile:0.1% TFA). The purity of each compound was confirmed with an analytical HPLC system using a Jupiter 10 µm C4 300 Å column (250 × 4.6 mm). The solvent composition was as follows: phase A (water:0.1% TFA) and phase B (acetonitrile:0.1% TFA); gradient, from 5% B to 95% B over a period of 15 min. The purity of all compounds was ≥95%. The molecular weight of each substrate was confirmed by high-resolution mass spectrometry using a Waters LCT premier XE with electrospray ionization and a time-of-flight module.
Enzyme preparation
Gene cloning and recombinant production of the SARS-CoV and SARS-CoV-2 Mpro are described elsewhere9,17.
Combinatorial library synthesis
Synthesis of H2N-ACC-resin
ACC synthesis was carried out according to Maly et al.28. To a glass reaction vessel, 1 eq. (9.62 mmol, 13 g) of Rink AM resin was added and stirred gently once per 10 min in DCM for 1 h, then filtered and washed three times with DMF. Fmoc-group deprotection was performed using 20% piperidine in DMF (three cycles of 5, 5 and 25 min), filtered and washed with DMF each time (six times). Next, 2.5 eq. of Fmoc-ACC-OH (24.05 mmol, 10.64 g) was preactivated with 2.5 eq. HOBt monohydrate (24.05 mmol, 3.61 g) and 2.5 eq. DICI (24.05 mmol, 3.75 ml) in DMF and the slurry was added to the resin. The reaction mixture was shaken gently for 24 h at room temperature. After this time, the resin was washed four times with DMF and the reaction was repeated using 1.5 eq. of above reagents to improve the yield of ACC coupling to the resin. After 24 h, the resin was washed with DMF and the Fmoc protecting group was removed using 20% piperidine in DMF (5, 5 and 25 min), filtered and washed with DMF (six times).
Synthesis of H2N-Gln(Trt)-ACC-resin
Here, 2.5 eq. Fmoc-Gln(Trt)-OH (24.05 mmol, 14.69 g) with 2.5 eq. HATU (24.05 mmol, 9.15 g) and 2.5 eq. collidine (24.05 mmol, 3.18 ml) in DMF were activated for 2 min and added to a filter cannula with 1 eq. (9.62 mmol) H2N-ACC-resin, and the reaction was carried out for 24 h. Next, the resin was washed four times with DMF and the same reaction was performed again using 1.5 eq. of these reagents. After four DMF washes, the Fmoc protecting group was removed using 20% piperidine in DMF (5, 5 and 25 min). Subsequently, the resin was washed with DCM (three times) and MeOH (three times) and dried over P2O5.
Library synthesis
The synthesis of P2, P3 and P4 sublibraries is exemplified in detail with the P2 sublibrary. The P2 library consisted of 137 compounds where all of the natural amino acids (omitting cysteine) and a pool of unnatural amino acids were used at a defined position (in this case, the P2 position) and an isokinetic mixture of 19 amino acids (without cysteine, plus norleucine mimicking methionine) was coupled in the remaining positions (in case of the P2 sublibrary, positions P3 and P4 were occupied by an isokinetic mixture). Equivalent ratios of amino acids in the isokinetic mixture were defined based on their reported coupling rates. A fivefold excess (over the resin load) of the mixture was used. For fixed positions, 2.5 eq. of single amino acid was used. All reactions were performed with the use of the coupling reagents DICI and HOBt. For P2 coupling, the synthesis of the library was performed using a MultiChem 48-well synthesis apparatus (FlexChem from SciGene). To each well of the reaction apparatus, 1 eq. of dry H2N-Gln(Trt)-ACC-resin (0.059 mmol, 80 mg) was added and stirred gently for 30 min in DCM, and then washed four times with DMF. In separate Eppendorf tubes, 2.5 eq. (0.15 mmol) Fmoc-P2-OH was preactivated with 2.5 eq. HOBt (0.15 mmol, 22.5 mg) and 2.5 eq. DICI (0.15 mmol, 23.55 μl) in DMF. Next, preactivated amino acids were added to wells of the apparatus containing H2N-Gln(Trt)-ACC-resin, followed by 3 h of agitation at room temperature. Then, the reaction mixture was filtered, washed with DMF (four times) and the ninhydrin test was carried out to confirm P2-amino-acid coupling. Subsequently, Fmoc protecting groups were removed with the use of 20% piperidine in DMF (5, 5 and 25 min). For P3 and P4 position coupling, an isokinetic mixture for 48 portions was prepared from 18 Fmoc-protected natural amino acids (omitting cysteine; plus norleucine mimicking methionine; 19 amino acids in total). Next, 5 eq. of isokinetic mixture, 5 eq. HOBt (14.16 mmol, 2.13 g) and 5 eq. DICI (14.16 mmol, 2.22 ml) were diluted in DMF and preactivated for 3 min. The activated isokinetic mixture was added to each of 48 wells containing 1 eq. of H2N-P2-Gln(Trt)-ACC-resin. After 3 h of gentle agitation, the slurry was filtered off and washed with DMF (four times). A ninhydrin test was carried out and the Fmoc protecting group was removed using 20% piperidine in DMF (5, 5 and 25 min). The same procedure was applied for the remaining compounds. The isokinetic mixture was added to prepare the P4 position in the same manner as for the P3 position. In the last step of the synthesis, N-terminal acetylation was performed; to prepare the mixture for 48 compounds, 5 eq. of AcOH (14.16 mmol, 807 µl), 5 eq. HBTU (14.16 mmol, 5.37 g) and 5 eq. DIPEA (14.16 mmol, 2.44 ml) in roughly 45 ml of DMF were added to a 50-ml falcon tube. After gentle stirring for 1 min, the mixture (roughly 800 µl) was added to each well in the reaction apparatus, containing the H2N-Mix-Mix-P2-Gln(Trt)-ACC-resin, followed by gentle agitation for 30 min. Next, the resin was washed six times with DMF, three times with DCM, three times with MeOH and dried over P2O5. After completing the synthesis, peptides were cleaved from the resin with a mixture of cold TFA:TIPS:H2O (%, v/v/v 95:2.5:2.5; 2 ml per well; 2 h, shaking once per 15 min). The solution from each well was collected separately and the resin was washed once with a portion of fresh cleavage solution (1 ml), followed by addition of diethyl ether (Et2O, 14 ml) into falcon tubes with peptides in solution. After precipitation (30 min at −20 °C), the mixture was centrifuged and washed again with Et2O (5 ml). After centrifugation, the supernatant was removed and the remaining white precipitate was dissolved in ACN/H2O (v/v, 3/1) and lyophilized. The products were dissolved in dimethylsulfoxide to a final concentration of 10 mM and used without further purification. The synthesis of P3 and P4 sublibraries was performed in the same manner as described above; P3 and P4 sublibraries were synthesized by coupling fixed amino-acid residues to the P3 (isokinetic mixture coupled to P2 and P4) and P4 positions (isokinetic mixture coupled to P2 and P3).
Library screening
HyCoSuL screening was performed using a spectrofluorometer (Molecular Devices Spectramax Gemini XPS) in 384-well plates (Corning). The assay conditions were as follows: 1 µl of substrate and 49 µl of enzyme, which was incubated at 37 °C for 10 min in assay buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.3). The final substrate concentration was 100 µM and the final enzyme concentration was 1 µM SARS-CoV and 0.6 µM SARS-CoV-2 Mpro, respectively. The release of ACC was measured for 45 min (excitation wavelength of 355 nm and emission wavelength of 460 nm) and the linear part of each progress curve was used to determine the substrate hydrolysis rate. Substrate specificity profiles were established by setting the highest value of relative fluorescence unit per second from each position as 100% and others were adjusted accordingly.
Kinetic analysis of substrates
Substrate screening was carried out in the same manner as the library assay. Substrate concentration was 5 µM, SARS-CoV Mpro was 0.3 µM and SARS-CoV-2 Mpro was 0.3 µM. Substrate hydrolysis was measured for 30 min using the following wavelengths: excitation, 355 nm and emission, 460 nm. The experiment was repeated three times. Results were presented as mean values with standard deviations. Kinetic parameters were assayed in 96-well plates (Corning). Wells contained 80 µl of enzyme in assay buffer (0.016–0.1 µM SARS-CoV-2 Mpro) and 20 µl of substrate at eight different concentrations ranging from 58.5 to 1,200 µM (0.44–7 µM for IQF substrates). ACC liberation was monitored for 30 min (excitation, 355 nm; emission, 460 nm). Each experiment was repeated at least three times. Kinetic parameters were determined using the Michaelis–Menten equation and GraphPad Prism software. Due to the precipitation of internally quenched fluorescence substrates in assay buffer, only the specificity constant (kcat/KM) was determined. When the [S0] << KM, the plot of vi (the initial velocities) versus [S0] yields a straight line with the slope representing Vmax/KM, kcat/KM = slope/E (E, total enzyme concentration).
Determination of inhibition kinetics (k obs/I) for inhibitors and ABPs
SARS-CoV-2 Mpro (75 nM) was preincubated in assay buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.3) for 10 min at 37 °C. Then, the enzyme was added to wells containing seven different concentrations of inhibitor or probe (ranging from 3.9 to 20 µM) and 50 μM of substrate (QS1). The measurement was conducted for 30 min and repeated at least three times; kobs/I was calculated as previously described21.
SARS-CoV-2 Mpro labeling
SARS-CoV-2 Mpro (5, 10, 50, 100 or 200 nM) was incubated with different probe concentrations (50–2,500 nM) in assay buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, pH 7.3) for 15 or 30 min at 37 °C. Then 3× SDS/DTT was added, and the samples were boiled for 5 min at 95 °C and resolved on 4–12% Bis-Tris Plus 12-well gels at 30 µl samples per well. Electrophoresis was performed at 200 V for 29 min. Next, the proteins were transferred to a nitrocellulose membrane (0.2 µm, Bio-Rad) for 60 min at 10 V. The membrane was blocked with 2% BSA in Tris-buffered saline with 0.1% (v/v) Tween 20 (TBS-T) for 60 min at room temperature. The biotinylated ABP was detected with a fluorescent streptavidin Alexa Fluor 647 conjugate (1:10,000) in TBS-T with 1% BSA using an Azure Biosystems Sapphire Biomolecular Imager (lasers of 658 and 488 nm for Bodipy FL dye) and Azure Spot Analysis Software. To perform a time-point analysis, SARS-CoV-2 Mpro (10 nM) was incubated with Bodipy-QS5-VS (2.5 µM) for different durations of time in the range from 1 to 60 min. Then the samples were boiled, and run on a gel. For electrophoresis, protein transfer to a nitrocellulose membrane and probe visualization were conducted in the same manner as described above.
SARS-CoV-2 Mpro labeling in cell lysates
HeLa and HEK-293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine and antibiotics (100 U ml−1 penicillin, 100 µg ml−1 streptomycin) in a humidified 5% CO2 atmosphere at 37 °C. Approximately 1,200,000 cells were collected and washed three times with PBS. The cell pellet was lysed in buffer containing 20 mM Tris, 150 mM NaCl and 5 mM DTT, pH 8.0, using a sonicator. The cell lysate was centrifuged for 10 min, and the supernatant was collected. Twenty microliters of cell lysate was incubated with or without 30 µl of inhibitor Ac-QS1-VS and with or without SARS-CoV-2 Mpro (100 or 25 nM) for 30 min at 37 °C. Next, 50 µl of probes at different concentrations was added to the samples and they were incubated for 15 or 30 min at 37 °C. Then the samples were combined with 50 µl 3× SDS/DTT, boiled and run on a gel. Electrophoresis, protein transfer to a nitrocellulose membrane and probe visualization were conducted in the same manner as described above. β-actin was used as a loading control (Source Data Fig. 1).
Antiviral activity of Ac-Abu-dTyr-Leu-Gln-VS toward a SARS-CoV-2 strain in Huh7 cell culture
The human hepatoma cell line Huh7 (kindly provided by R. Bartenschlager, University of Heidelberg, Germany) was maintained in DMEM (Gibco catalog no. 41965-039) supplemented with 10% fetal bovine serum (FBS), 2% HEPES 1 M (Gibco catalog no. 15630106), 5 ml of sodium bicarbonate 7.5% (Gibco catalog no. 25080-060) 1% nonessential amino acids (NEAA Gibco catalog no. 11140050) and 1% penicillin-streptomycin 10,000 U ml−1 (Gibco catalog no. 15140148) in a humidified 5% CO2 incubator at 37 °C. Assay medium, used for producing virus stocks and antiviral testing, was prepared by supplementing DMEM with 4% FBS, 2% HEPES 1 M, 5 ml of sodium bicarbonate 7.5 and 1% NEAA.
To quantify antiviral activity on Huh7 cells, we selected a SARS-CoV-2 virus strain that produces sufficient cytopathogenic effect (CPE) on this cell line. We started from passage 6 of the SARS-CoV-2 strain BetaCov/Belgium/GHB-03021/2020 (EPI ISL 407976, 3 February 2020) that has been described previously29, and passaged this three additional times on Huh7 cells while selecting those cultures that showed most CPE. This resulted in a virus stock (passage 9) that confers full CPE on Huh7 (5.6 × 104 median tissue culture infectious dose (TCID50) per ml) as well as on VeroE6 cells (1.8 × 107 TCID50 per ml). The genotype of this virus stock shows four nucleotide changes as compared with the mother virus stock (P6) and these are currently being analyzed. None of the nucleotide changes occur in the part of the genome that encodes the 3C-like protease, validating this virus stock for testing protease inhibitors.
For antiviral testing, Huh7 cells were seeded in 96-well plates (Corning CellBIND 96-well Microplate catalog no. 3300) at a density of 6,000 cells per well in assay medium. After overnight growth, cells were treated with the indicated compound concentrations and infected with a multiplicity of infection of 0.005 TCID50 per cell of the P9 virus (final volume of 200 μl per well in assay medium). On day 4 postinfection, differences in cell viability caused by virus-induced CPE or by compound-specific side effects were analyzed using MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt). For this, an MTS:phenazine methosulfate (PMS) stock solution (2 mg ml−1 MTS (Promega) and 46 µg ml−1 PMS (Sigma-Aldrich) in PBS at pH 6–6.5) was diluted 1:20 in MEM without phenol red (Gibco catalog no. 51200038). Medium was aspirated from wells of the test plates and 70 µl of MTS/PMS solution was added. After 0.5–1 h incubation at 37 °C, absorbance was measured at 498 nm. Cytotoxic effects caused by compound treatment alone were monitored in parallel plates containing mock-infected cells.
All virus-related work was conducted in the high-containment BSL3+ facilities of the KU Leuven Rega Institute (3CAPS) under licenses AMV 30112018 SBB 219 2018 0892 and AMV 23102017 SBB 219 2017 0589 according to institutional guidelines.
Crystallization of SARS-CoV-2 Mpro in complex with Biotin-PEG(4)-Abu-Tle-Leu-Gln-VS
The purified SARS-CoV-2 Mpro was concentrated to 23 mg ml−1, mixed with Biotin-PEG(4)-Abu-Tle-Leu-Gln-VS at a molar ratio of 1:5 and the mixture was incubated at 4 °C overnight. The next day, the mixture was clarified by centrifugation at 12,000g, 4 °C and the supernatant was set for crystallization screening by using commercially available screening kits (PEGRxT 1 and 2 (Hampton Research) and Morpheus HT-96 (Molecular Dimensions)). A Gryphon LCP crystallization robot (Art Robbins) was used for setting up the crystallization screens with the sitting-drop vapor-diffusion method at 18 °C, where 0.15 μl of protein solution and 0.15 μl of reservoir were mixed to equilibrate against 40 μl of reservoir solution.
Crystals were observed under several conditions in the two 96-well plates. The crystals were fished directly from the basic screening plates. The cryo-protectant consisted of mother liquor plus varied concentrations (5 to 20%) of glycerol, and 2 mM of the ABP. Subsequently, liquid nitrogen was used for flash-cooling the crystals before data collection.
Diffraction data collection and determination of the structure
Several diffraction datasets were collected at 100 K at the P11 beamline of PETRA III (DESY), using synchrotron radiation of wavelength 1.0332 Å and a Pilatus 6M detector (Dectris). For structure determination, a dataset was used that was collected using a crystal fished from the condition no. E7 of Morpheus HT-96 (0.12 M ethylene glycols (0.3 M diethylene glycol, 0.3 M triethylene glycol, 0.3 M tetraethylene glycol, 0.3 M pentaethylene glycol), 0.1 M buffer system 2 (1.0 M sodium HEPES, MOPS (acid), pH 7.5), pH 7.5, 30% precipitant mix 3 (20% v/v glycerol, 10% PEG 4000)).
XDSapp30, Pointless31,32 and Scala31 (the last two from the CCP4 suite33) were used for processing and scaling the dataset. The space group was determined as P6122 and the resolution limit was set at a Bragg spacing of 1.70 Å. The molecular replacement method was used for phase determination using the Molrep program33,34 and the free-enzyme crystal structure of SARS-CoV-2 Mpro (PDB 6Y2E, ref. 9) as the search model. The geometric restraints for the ABP were generated using the Jligand programe from the CCP4 suite33,35; the ABP was built into the Fo–Fc density by using the Coot software36. Structure refinement was performed with Refmac5 (refs. 33,36,37). Ramachandran statistics after structure refinement are as follows: preferred region 97.0%, allowed region 3.0% and outlier region 0.0%. Statistics of diffraction data processing and model refinement are shown in Supplementary Table 4.
Patient-sample preparation
The study was approved by the Bioethics Committee of the Medical University of Lodz, Poland (no. RNN/114/20/KE). The study was conducted in compliance with good clinical practice guidelines and under the principles of the Declaration of Helsinki. The study participants or their parents provided written informed consent. Patients were hospitalized at the Central Medical University Hospital in Lodz (CUHL), Poland and from the Department of Infectious Diseases and Hepatology, Medical University of Lodz (DIDH), Poland, and were positive for RNA of SARS-CoV-2 in serial measurements of nasopharyngeal swabs (at least two consecutive measurements) with clinical symptoms of infection.
For the current study, two nasopharyngeal swabs were collected from each patient at the same time, one was subjected to RNA SARS-CoV-2 testing and the other was used for smear preparation for confocal laser scanning microscopy. Smears were performed on glass slides covered with polylysine (Thermo Fisher Scientific).
Clinical characteristics of one patient (P01) recruited at CUHL
P01, a 13-year old boy, presented with mild symptoms of COVID-19 infection and no need for respiration support or mechanical ventilation; no specific treatment was applied. Two nasopharyngeal swabs for Mpro activity were collected during routine clinical examination, at days 1 and 5 after COVID-19 diagnosis. Tests positive for SARS-CoV-2 RNA were obtained on days 1 and 5, the first negative test was on day 7.
Clinical characteristics of six patients recruited at DIDH
P01DZ, a 59-year old woman, presented with moderate symptoms of COVID-19 infection, was hospitalized on day 3 after diagnosis, treated with respiration support (oxygen 3 l min−1, for 10 d) and no mechanical ventilation was needed. She was treated with dexamethasone, hydroxychloroquine, lopinavir and ritonavir. One nasopharyngeal swab for Mpro activity was collected on day 12 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1, 3, 12 and the first negative test was on day 20.
P02JB, a 34-year old woman, presented with moderate to severe COVID-19 pneumonia, was hospitalized on day 1 after diagnosis, treated with respiration support (oxygen 3 l min−1, for 20 d) and no mechanical ventilation was needed. She was treated with dexamethasone, hydroxychloroquine, remdesivir and vancomycin because of concomitant Clostridium difficile infection. One nasopharyngeal swab for Mpro activity was collected on day 10 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1, 7, 10 and 14, the first negative test was obtained on day 21.
P03KB, a 36-year old man, presented with moderate symptoms of COVID-19 pneumonia, was hospitalized on day 1 after diagnosis, treated with respiration support (oxygen 3 l min−1, for 6 d) and no mechanical ventilation was needed. He was treated with dexamethasone, hydroxychloroquine, lopinavir and ritonavir. One nasopharyngeal swab for Mpro activity was collected on day 10 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1, 7, 10 and the first negative test was on day 12.
P04WA, a 69-year old woman, presented with moderate symptoms of COVID-19 pneumonia, was hospitalized on day 1 after diagnosis, with respiration support (oxygen 3 l min−1, required at the time of article submission). She was treated with hydroxychloroquine, dexamethasone and ceftriaxone. One nasopharyngeal swab for Mpro activity was collected on day 5 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1 and 5.
P05WM, a 72-year old man, presented with moderate symptoms of COVID-19 pneumonia, was hospitalized on day 1 after diagnosis, with respiration support (oxygen 3 l min−1, required at the time of article submission). He was treated with hydroxychloroquine and dexamethasone. One nasopharyngeal swab for Mpro activity was collected on day 5 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1 and 5.
P06JM, a 34-year old man with moderate symptoms of COVID-19 pneumonia, was hospitalized on day 1 after diagnosis, no needs for respiration support or mechanical ventilation. He was treated with hydroxychloroquine and ceftriaxone. One nasopharyngeal swab for Mpro activity was collected on day 5 after COVID-19 diagnosis during routine clinical examination. Tests for SARS-CoV-2 RNA were positive on days 1 and 5.
Detection of SARS-CoV-2 Mpro in epithelial cells by immunofluorescence
The nasopharyngeal swab was used for smear preparation for confocal laser scanning microscopy. Smears were performed on glass slides covered with polylysine (Thermo Fisher Scientific). These slides were treated with 1 or 2.5 μM probe of Mpro (Cy5-QS1-VS or Bodipy-QS5-VS) and incubated for 30 min at 37 °C. Slides were fixed in 70% ethanol for 30 min. For further staining overnight at 4 °C, incubation with recombinant goat anti-ACE2 antibody (R&D Systems, 1:20) was performed. The next day, antibodies were aspirated, and cells were washed twice with PBS and labeled with a secondary antibody (Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 (catalog no. A32814, 1:200 Invitrogen, Thermo Fisher Scientific) or Cy5 AffiniPure donkey anti-Goat IgG (H+L) (705-175-147, 1:200, Jackson ImmunoResearch)) in PBS for 1 h at room temperature. Next, the cells were washed twice with PBS, coverslips were mounted with Vectashield fluorescence mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratory H-1000) and sealed with nail polish. Slides were stored at 4 °C until use. Cells were then subjected to confocal microscopy analysis using a Leica TCS SP8. DAPI was detected by the DAPI channel (405 nm), the Mpro Cy5-QS1-VS probe was detected with the Cy5 filter single photon laser (638 nm) (Bodipy-QS5-VS 488 nm) and ACE2/secondary antibodies were read using the fluorescein isothiocyanate filter (single photon laser 458 nm) or Cy5 filter laser 638 nm. All images were acquired in .tiff format using Leica Application Suite X software. Images shown are representative views of cells from two coverslips.
Slides with inhibitor (Ac-QS1-VS) were prepared in the same manner as described above. Cells were treated with 50 μM Ac-QS1-VS and incubated for 30 min at 37 °C. Then cells were washed twice with PBS and incubated with 2.5 μM Bodipy-QS5-VS for 30 min at 37 °C.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information files. The expression plasmid for the SARS-CoV-2 main protease is available from R.H. (rolf.hilgenfeld@uni-luebeck.de). The atomic coordinates and structure factors for the complex between the SARS-CoV-2 Mpro and the ABP have been deposited in the Protein Data Bank under code 6Z2E. Source data are provided with this paper.
References
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473 (2020).
Huang, C. L. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Jiang, S. B. et al. A distinct name is needed for the new coronavirus. Lancet 395, 949–949 (2020).
Coronavirus Disease (COVID-2019) Situation Reports, 5 August 2020 https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200805-covid-19-sitrep-198.pdf?sfvrsn=f99d1754_2 (WHO, 2020).
Li, G. D. & De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 19, 149–150 (2020).
Hilgenfeld, R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096 (2014).
Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y. & Jung, S. H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 59, 6595–6628 (2016).
Zhang, L. L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 (2020).
Wu, A. P. et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CL(pro)) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Wu, A. D. et al. Prediction and biochemical analysis of putative cleavage sites of the 3C-like protease of Middle East respiratory syndrome coronavirus. Virus Res. 208, 56–65 (2015).
Chuck, C. P., Chow, H. F., Wan, D. C. C. & Wong, K. B. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PloS ONE 6, https://doi.org/10.1371/journal.pone.0027228 (2011).
Zhang, L. L. et al. alpha-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 63, 4562–4578 (2020).
Yang, H. T. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA 100, 13190–13195 (2003).
Zhu, L. L. et al. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antivir. Res. 92, 204–212 (2011).
Jin, Z. M. et al. Structure of M-pro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Aleshin, A. E. et al. Activity, specificity, and probe design for the smallpox virus protease K7L. J. Biol. Chem. 287, 39470–39479 (2012).
Poreba, M. et al. Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 21, 1482–1492 (2014).
Kasperkiewicz, P. et al. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl Acad. Sci. USA 111, 2518–2523 (2014).
Kasperkiewicz, P., Altman, Y., D’Angelo, M., Salvesen, G. S. & Drag, M. Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J. Am. Chem. Soc. 139, 10115–10125 (2017).
Rut, W., Poreba, M., Kasperkiewicz, P., Snipas, S. J. & Drag, M. Selective substrates and activity-based probes for imaging of the human constitutive 20S proteasome in cells and blood samples. J. Med. Chem. 61, 5222–5234 (2018).
Poreba, M. et al. Selective imaging of cathepsin L in breast cancer by fluorescent activity-based probes. Chem. Sci. 9, 2113–2129 (2018).
Poreba, M. et al. Small molecule active site directed tools for studying human caspases. Chem. Rev. 115, 12546–12629 (2015).
Maluch, I., Czarna, J. & Drag, M. Applications of unnatural amino acids in protease probes. Chem.- Asian J. 14, 4103–4113 (2019).
Poreba, M. et al. Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells. Cell Death Differ. 26, 2695–2709 (2019).
Maly, D. J. et al. Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J. Org. Chem. 67, 910–915 (2002).
Boudewijns, R. et al. STAT2 signaling as double-edged sword restricting viral dissemination but driving severe pneumonia in SARS-CoV-2 infected hamsters. Preprint at bioRxiv https://doi.org/10.1101/2020.04.23.056838 (2020).
Krug, M., Weiss, M. S., Heinemann, U. & Mueller, U. XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 45, 568–572 (2012).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D. Struct. Biol. 62, 72–82 (2006).
Evans, P. R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D. Biol. Crystallogr. 67, 282–292 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Struct. Biol. 67, 235–242 (2011).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25 (2010).
Lebedev, A. A. et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D. Struct. Biol. 68, 431–440 (2012).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Struct. Biol. 67, 355–367 (2011).
Acknowledgements
The Drag laboratory is supported by the National Science Center in Poland and the ‘TEAM/2017-4/32’ project, which is conducted within the TEAM program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund. Work in the Hilgenfeld laboratory was supported by the SCORE project of the European Union (grant agreement no. 101003627), by DZIF and by the Government of Schleswig-Holstein through its Structure and Excellence Fund, as well as by a close partnership between the Possehl Foundation (Lübeck) and the University of Lübeck. Work in the Neyts laboratory was similarly supported by the SCORE project of the European Union (grant agreement no. 101003627). X.W. received funding of the China Scholarship Council (grant no. 201806170087). L.Z. is supported by a stipend from DZIF. W.R. is a beneficiary of a START scholarship from the Foundation for Polish Science. The Mlynarski laboratory (B.P. and W.M.) is supported by the FIXNET project, which is conducted within the TEAM-NET program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund (no POIR.04.04.00-00-1603/18). We thank A. Piekarska (Medical University of Lodz, Poland) for an expert consultation in clinical assessments of patients with COVID-19.
Author information
Authors and Affiliations
Contributions
M.D. and W.R. designed the research. W.R., K.G., L.Z., X.W., D.J. and M.Z. performed the research and collected data. R.H., X.S. and L.Z. contributed enzymes. L.Z. and R.H. solved and refined the crystal structure. B.P. and W.M. carried out labeling experiments in patient samples and confocal imaging. W.R., L.Z., W.M., X.W., D.J., J.N., R.H. and M.D. analyzed and interpreted the data. W.R., L.Z., R.H. and M.D. wrote the paper. All authors critically revised the paper.
Corresponding authors
Ethics declarations
Competing interests
Wroclaw University of Science and Technology has filed a patent application covering the compounds Ac-Abu-Tle-Leu-Gln-VS, Ac-Abu-dTyr-Leu-Gln-VS, Biotin-PEG(4)-Abu-Tle-Leu-Gln-VS and Cy5-PEG(4)-Abu-Tle-Leu-Gln-VS as well as related compounds with W.R. and M.D. as inventors.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The general library structure.
Structure of HyCoSuL library designed for P1-Gln-specific endopeptidases.
Extended Data Fig. 2 The rate of substrate hydrolysis by SARS-CoV Mpro and SARS-CoV-2 Mpro.
[S]=5 µM, [E]=0.3 µM; n=3, where n represents the number of independent experiments. Error bars represent mean ± SD.
Extended Data Fig. 3 Kinetic measurements of SARS-CoV-2 Mpro substrates.
The measurements were performed in three independent replicates. Data are presented as mean values +/- SD. Due to internally quenched fluorescence substrate precipitation (IQF QS1, QS2 and QS4) in assay buffer only specificity constant could be determined.
Extended Data Fig. 4 SARS-CoV-2 Mpro detection by activity-based probes.
a, Structure of inhibitor and activity-based probes. b, SARS-CoV-2 Mpro labelling by probes (B-QS1-VS and Cy5-QS1-VS). c, SARS-CoV-2 Mpro probe selectivity in HeLa lysate (asterisk shows SARS-CoV-2 Mpro band, which was spiked into the cell lysate). The cell lysate was incubated with or without the inhibitor Ac-QS1-VS for 30 min at 37 °C; next, the different probe concentrations were added and the samples were incubated for 15 min at 37 °C. The biotinylated activity-based probe was detected with a fluorescent streptavidin Alexa Fluor 647 conjugate (1:10,000) in TBS-T with 1% BSA using an Azure Biosystems Sapphire Biomolecular Imager and Azure Spot Analysis Software. The last lane on the membranes is HeLa lysate only. Each experiment was repeated independently two times with similar results.
Extended Data Fig. 5 Kinetic measurement of SARS-CoV-2 Mpro inhibitors and activity-based probes.
Product formation (P) proceeded at an initial velocity (V) and was inhibited over time (t) at a rate of (kapp): P=V/kapp*(1-e-kapp*t)+C (kapp− is the apparent rate of inhibition in the presence of substrate). kapp was determined separately for each inhibitor/probe concentration using non-linear regression analysis in GraphPad Prism software. The obtained values were plotted against inhibitor/probe concentration giving kapp/I (apparent second order rate constant). kobs/I (absolute value of the second order rate constant) was calculated from kobs/I = kapp/I * (1+[S]/KM). The measurements were performed in three independent replicates. Data are presented as mean values +/- SD.
Extended Data Fig. 6 Three-dimensional structure of the activity-based probe (ABP) Biotin-PEG(4)-Abu-Tle-Leu-Gln-VS (B-QS1-VS) in complex with the SARS-CoV-2 Mpro.
a, In the crystal, the ABP links two neighboring Mpro dimers. The vinylsulfone warhead and the P1 - P4 residues bind to the substrate-binding site of the parent Mpro dimer (wheat), while the PEG(4) unit and the biotin label are in contact with the neighboring Mpro dimer (grey). b, At the monomer - monomer interface of the Mpro, a chloride ion from the crystallization buffer occupies a position on the two-fold axis.
Extended Data Fig. 7 SARS-CoV-2 Mpro detection by the activity-based probe in nasopharyngeal epithelial cells from COVID-19 patients positive for SARS-CoV-2 RNA (P01).
Confocal microscopy of the epithelial cells of nasopharyngeal swabs co-stained with the Cy5-QS1-VS SARS-CoV-2 Mpro probe, anti-ACE2 antibody (ACE2 - angiotensin-converting enzyme 2) with Alexa Fluor 488 donkey anti-Goat secondary antibody, and DAPI (4′,6-diamidino-2-phenylindole) in patient P01 at days 1 and 5 after diagnosis as well as in a healthy control. Due to ethical concerns one nasopharyngeal swab was collected from patient P01 on day 1 and day 5.
Extended Data Fig. 8 SARS-CoV-2 Mpro detection by the activity-based probe (Bodipy-QS5-VS) in nasopharyngeal epithelial cells from patients positive (panel a, b) and negative for SARS-CoV-2 RNA (CTRL 1-3, panel c).
Confocal microscopy of the epithelial cells of nasopharyngeal swabs co-stained with Bodipy-QS5-VS SARS-CoV-2 Mpro probe and anti-ACE2 antibody with Cy5 AffiniPure donkey anti-Goat secondary antibody and DAPI. Panel b - cells were treated with 50 µM Ac-QS1-VS and incubated for 30 min at 37 °C. Then cells were washed twice with PBS and incubated with 2.5 µM Bodipy-QS5-VS for 30 min at 37 °C. Due to ethical concerns one nasopharyngeal swab was collected from each patient.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and note.
Source data
Source Data Fig. 1
Full-length scan of membranes.
Rights and permissions
About this article

Cite this article
Rut, W., Groborz, K., Zhang, L. et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat Chem Biol 17, 222–228 (2021). https://doi.org/10.1038/s41589-020-00689-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-020-00689-z
This article is cited by
-
Preclinical evaluation of the SARS-CoV-2 Mpro inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir
Nature Microbiology (2024)
-
Therapeutic strategies for COVID-19: progress and lessons learned
Nature Reviews Drug Discovery (2023)
-
Ebselen derivatives inhibit SARS-CoV-2 replication by inhibition of its essential proteins: PLpro and Mpro proteases, and nsp14 guanine N7-methyltransferase
Scientific Reports (2023)
-
Calpeptin is a potent cathepsin inhibitor and drug candidate for SARS-CoV-2 infections
Communications Biology (2023)
-
Predicted coronavirus Nsp5 protease cleavage sites in the human proteome
BMC Genomic Data (2022)
