
Abstract
Neutralizing agents against SARS-CoV-2 are urgently needed for the treatment and prophylaxis of COVID-19. Here, we present a strategy to rapidly identify and assemble synthetic human variable heavy (VH) domains toward neutralizing epitopes. We constructed a VH-phage library and targeted the angiotensin-converting enzyme 2 (ACE2) binding interface of the SARS-CoV-2 Spike receptor-binding domain (Spike-RBD). Using a masked selection approach, we identified VH binders to two non-overlapping epitopes and further assembled these into multivalent and bi-paratopic formats. These VH constructs showed increased affinity to Spike (up to 600-fold) and neutralization potency (up to 1,400-fold) on pseudotyped SARS-CoV-2 virus when compared to standalone VH domains. The most potent binder, a trivalent VH, neutralized authentic SARS-CoV-2 with a half-maximal inhibitory concentration (IC50) of 4.0 nM (180 ng ml−1). A cryo-EM structure of the trivalent VH bound to Spike shows each VH domain engaging an RBD at the ACE2 binding site, confirming our original design strategy.

Similar content being viewed by others


Main
The emergence of SARS-CoV-2 and the associated COVID-19 disease has emphasized the need to rapidly generate therapeutics against novel pathogens. SARS-CoV-2 enters cells through the interaction of the viral Spike receptor-binding domain (Spike-RBD) and host angiotensin-converting enzyme-2 (ACE2) on the surface of lung epithelial cells1. Antibody and antibody-like biologics that can block this interaction are promising therapeutic candidates because of their high specificity and neutralization potency2. The majority of antibodies isolated so far against SARS-CoV-2, SARS-CoV-1 and MERS are derived from screening the B cells of infected patients or repurposed from animal immunizations3,4,5,6,7. These approaches, though effective, can be time-consuming and may not necessarily yield neutralizing antibodies. Given the pressing nature of this pandemic, there is a need for multiple parallel strategies to rapidly produce potent, recombinant and neutralizing biologics.
In vitro display technologies using yeast or phage are well-established approaches for generating high-affinity binders from large naive libraries8. In vitro selection can be done without infected individuals and only requires the recombinant protein target. One of the recently developed modalities comprises small single-domain antibodies derived from variable heavy homodimer (VHH) domains of antibodies from camels or llamas, often referred to as nanobodies, and are usually obtained by camelid immunization and B-cell cloning9,10,11,12. Nanobodies have some advantages. Their single chain and small size (11–15 kDa) allow them to bind epitopes or penetrate tissues that may not be accessible to monoclonal antibodies (mAbs; 150 kDa), and nanobodies can be produced rapidly in Escherichia coli13,14. However, nanobodies derived from animal immunization can also suffer from long turnaround times. Although this can be overcome with synthetic nanobody libraries15,16, nanobody scaffolds that are animal-derived raise significant concerns regarding immunogenicity. More recently, variable heavy (VH) domains derived from human scaffolds have been produced and tested against a number of targets17,18,19.
Accordingly, we and others have been interested in developing VH binders to SARS-CoV-2 for the present pandemic, and as a test case for future ones6,20,21,22. However, one limitation of synthetic single-domain binders is that, as monomers, they often lack the strong binding affinity necessary for therapeutic application. Affinity maturation can improve this, although this extends the development timeline. Instead, generating linked multivalent or multi-paratopic VH binders could be a more rapid approach to utilize avidity to boost affinity and efficacy23. Linking VH domains into such homo- and hetero-bifunctional formats is more straightforward than preparing similar multifunctional antibodies, because the latter requires correct heavy- and light-chain pairing to maintain binding affinity, whereas multifunctional VH domains have no such requirements11.
Here, we construct a human VH-phage library derived from the clinically approved trastuzumab scaffold and validate its use on multiple antigens. By utilizing a masked phage selection strategy, we rapidly identify VH domains at two non-overlapping epitopes within the ACE2 binding site of the SARS-CoV-2 Spike-RBD. By linking these VH domains with a strategic linker into bi-paratopic and multivalent binders, we improve the affinity from mid-nanomolar to ~100 pM, without any additional high-resolution structural information. These high-affinity binders are capable of potently neutralizing pseudotyped and live SARS-CoV-2. A cryo-EM structure of the most potent trivalent VH bound to Spike shows that each VH domain precisely targets the ACE2 binding interface on all three RBDs of Spike. We believe our VH-phage library and this multivalent and multi-paratopic approach is highly advantageous in targeting distinct epitopes within an antigen and can be broadly applied to other viral and non-viral targets to leverage avidity for increased potency.
Results
Construction of a synthetic human VH-phage library
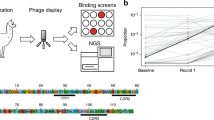
To enable the generation of single-domain antibodies against targets such as SARS-CoV-2 Spike, we designed a synthetic VH-phage library using the VH domain (4D5) from the highly stable and clinically successful trastuzumab antibody (Fig. 1a)24,25. The VH scaffold was modified to include five amino-acid changes predicted to reduce aggregation (Supplementary Table 1)26. To bias toward colloidal stability, aspartate and arginine or glutamate residues were inserted at the beginning and at two or three terminal positions of the complementarity-determining region (CDR) H1, as these have been used previously to improve the aggregation resistance of VH and scFv fragments from the VH3 germline19,27. Diversity was introduced into CDR H1 and CDR H2 using a minimalistic approach, where variability was largely restricted to tyrosine and serine residues (Extended Data Fig. 1)28. We introduced high-diversity mixtures of amino acids into CDR H3 because it is usually critical to antigen recognition (Extended Data Fig. 1), and Fab-phage libraries with highly diverse CDR H3 sequences have successfully yielded high-affinity antibodies to a variety of target antigens29,30. Furthermore, charged polar residues such as aspartate were introduced at 10% frequency to decrease the net surface hydrophobicity to mitigate aggregation and decrease the propensity for non-specific binders in the library.

a, Three-dimensional (3D) surface representation (left) of the VH-4D5 parental scaffold (PDB 1FVC) and a cartoon diagram (right), where individual CDRs are annotated in color with the designed loop length variations according to the Kabat nomenclature31. b, NGS analysis of the longest H3 loop (X = 16), showing that the expected global amino-acid frequencies are comparable to the designed frequencies. The gray shaded region denotes the 95% confidence interval. c, Representative NGS analysis of the longest H3 loop (X = 16), showing that the positional frequency distribution matches the designed frequencies. Position 1 refers to residue 95 (Kabat definition). Data for the other CDR H3 lengths are reported in Supplementary Fig. 2. d, NGS analysis of unique clones, showing that all H3 lengths are represented in the pooled VH-phage library.
Based on previous designs, we chose loop length variations of 5–7 residues in CDR H1 and 6–20 residues in CDR H3, while CDR H2 was kept constant at 17 residues (Kabat definitions; Extended Data Fig. 1 and Supplementary Table 2)29,31. To cover this large sequence space with a minimal bias towards different length variants, five separate sub-libraries were constructed by binning CDR H3 loop length insertions (X2–16) in incremental sets of three and combined to yield a final library of ~5 × 1010 transformants (Supplementary Fig. 1). Analysis of the unique CDR H3 sequences by next-generation sequencing (NGS) showed that the observed amino-acid frequencies closely matched our designs and all CDR H3 length variants were represented in the final library (Fig. 1b–d and Supplementary Fig. 2). Finally, to test the performance of the library, several rounds of panning were performed on representative antigens including both cytosolic and membrane proteins. These panning experiments were done in parallel with an in-house Fab-phage library. For all target antigens, high levels of enrichment were observed (Supplementary Fig. 3). For the majority of antigens, enrichment levels were comparable or substantially higher for the VH-phage library than for the Fab-phage library.
Identification of VHs that target multiple epitopes
So far, most neutralizing mAbs against SARS-CoV-2 target Spike and, not surprisingly, many of the most potent target the ACE2 binding interface3,7. Although cryo-EM structures show that the ACE2 binding interface remains largely solvent-accessible in both the RBD ‘up’ and ‘down’ conformations32, simultaneous intra-molecular engagement by both binding arms of mAbs may be challenging as they are not geometrically arranged to engage multiple RBDs on a single Spike trimer. Thus, our goal was to target this highly neutralizing epitope with VHs and subsequently link them together to utilize avidity beyond that of a homo-bivalent mAb.
We first expressed the Spike-RBD (residues 328–533) and the ACE2 peptidase domain (residues 1–640) as biotinylated Fc-fusions for VH-phage selections33. To specifically enrich for VH-phage that bind the ACE2 binding site on Spike-RBD, the library was first cleared with the Spike-RBD-Fc/ACE2-Fc complex to remove phage that bound outside the ACE2 binding interface. This was followed by selection on Spike-RBD-Fc alone to enrich for phage that bound the unmasked ACE2 binding site (Fig. 2a). By rounds 3 and 4, significant enrichments for phage that bound Spike-RBD-Fc but not the Spike-RBD-Fc/ACE2-Fc complex were observed (Supplementary Fig. 4a). Single clones were isolated and characterized for their ability to bind Spike-RBD-Fc by phage enzyme-linked immunosorbent assays (ELISAs; Supplementary Fig. 4b). Nearly all VH-phage showed enhanced binding to Spike-RBD-Fc over the Spike-RBD-Fc/ACE2-Fc complex, suggesting they bound the same epitope as ACE2 and could potentially block this interaction (Supplementary Fig. 4c). In total, 85 unique VH-phage sequences were identified, and a subset was characterized as recombinant VH domains. We identified three lead VH candidates that bound Spike-RBD with Kd values ranging from 23 to 113 nM (Fig. 2b–d). These VH were specific to Spike-RBD and did not recognize other in-house antigens (Extended Data Fig. 2). Epitope binning demonstrated that the three VH domains mapped to two non-overlapping epitopes (which we call site A and site B) within the larger ACE2 binding site (Supplementary Fig. 5). The VH domain that recognizes site A (A01) binds independently from the VHs that recognize site B (B01 and B02) (Fig. 2e–h).

a, Diagram illustrating the phage selection strategy to isolate VH-phage that bind at the ACE2 binding interface. Red indicates clearance of the phage pool by the Spike-RBD-Fc/ACE2-Fc complex; green indicates positive selection against Spike-RBD-Fc alone. To increase stringency, successively lower concentrations of Spike-RBD-Fc were used and, after four rounds of selection, individual phage clones were analyzed by phage ELISA. b–d, Bio-layer interferometry (BLI) of VH A01 (b), VH B01 (c) and VH B02 (d) against Spike-RBD. e–g, BLI-based epitope binning of VH A01 and VH B01 (e), VH A01 and VH B02 (f) and VH B01 and VH B02 (g). The antigen loaded onto the sensor tip was Spike-RBD. h, Diagram of the two different epitope bins targeted by VH domains.
Bi-paratopic and multivalent linkage increases affinity
We chose two parallel approaches to increase the affinity of the VH binders to Spike through avidity. First, we reasoned that VHs targeting site A or site B are in close proximity because they are non-overlapping but compete for the larger ACE2 binding site. Therefore, these VHs could be linked together to engage the same RBD simultaneously and improve affinity through intra-RBD avidity. Using the three VH monomers (A01, B01, B02) as modular units, we generated two bi-paratopic linked dimers (VH2) by fusing A01 with B01 or B02 (Fig. 3a). In a parallel approach, we aimed to leverage the trimeric nature of Spike and engage multiple RBDs on the same Spike simultaneously to improve affinity through inter-RBD avidity. To that end, we generated mono-paratopic Fc fusions (VH-Fc), linked dimers (VH2) and linked trimers (VH3) (Fig. 3a). The VH2 and VH3 consisted of a C-to-N terminal fusion of two or three VH monomers via a 20-amino-acid Gly-Ser linker (~70 Å), while the VH-Fc consisted of a fusion of VH to the human IgG1 Fc domain via a flexible Fc hinge (~100 Å). The structure of the SARS-CoV-2 Spike trimer suggests that the linked VH domains could bridge the distance between RBDs on an individual Spike (<55 Å), but are unlikely to span RBDs between discrete Spike proteins based on the inter-Spike distance on the viral envelope (150–180 Å) (Extended Data Fig. 3)32,34.

a, Cartoon depiction of engineered VH binders generated by linking VH domains via Fc-fusion or a 20-amino-acid Gly-Ser linker. b–d, BLI traces of lead VH binders VH-Fc B01 (b), VH2 A01-B01 (c) and VH3 B01 (d) against RBD (upper panel) or Secto (lower panel). e, Sequential BLI binding experiments measuring binding of ACE2-Fc to Secto pre-blocked with our VH binders, showing that multivalent VH binders can block ACE2-Fc binding to Secto. f, Competition serology ELISA with convalescent patient sera, showing that VH-Fc binders can compete with patient antibodies. P1 to P9 represent sera from individuals with a history of previous SARS-CoV-2 infection. C1 and C2 are sera collected from two donors before the SARS-CoV-2 outbreak. Individual data points represent technical replicates (n = 2) from the serum of the same individual and are shown as black circles.
ELISA and bio-layer interferometry (BLI) binding assays to Spike-RBD show that VH-Fc, VH2 and VH3 have 2.7- to 600-fold higher affinity to Spike-RBD (Kd = 0.1–8.4 nM) than the standalone VH monomers (Fig. 3b–d, Extended Data Fig. 4, Supplementary Tables 3 and 4, and Supplementary Fig. 6). Interestingly, fold increases in affinity were greater for binders that target site B or both site A and site B combined. The most potent constructs bound trimeric Spike ectodomain (Secto) with Kd values in the hundreds of picomolar range, and all utilized VH B01 (Fig. 3b–d). Next, we examined whether these multivalent VH can block ACE2 binding to Spike by testing several high-affinity constructs (VH-Fcs; VH2 A01-B01, VH2 A01 and VH2 B02) in a sequential BLI binding assay. Secto was immobilized on the biosensor, pre-blocked with each VH binder, and then assayed for binding to ACE2-Fc (Fig. 3e). We found that all tested binders substantially blocked binding of ACE2-Fc to Secto. Similarly, we examined whether these engineered VH-Fcs can compete with SARS-CoV-2 Spike-reactive antibodies in convalescent patient serum. Using a competition ELISA format previously developed by our group35, we found that VH-Fcs reduced the binding of patient antibodies to SARS-CoV-2 Spike-RBD (Fig. 3f). Taken together, these data show that modular reformatting of these VH domains can significantly increase the affinity to Spike and block the same immunogenic epitopes as patient-derived Abs.
Finally, we characterized the biophysical properties of these engineered VH by differential scanning fluorimetry (DSF), size exclusion chromatography (SEC) and reconstitution after lyophilization. The VH binders can be expressed in E. coli in high yields (VH2 A01-B01 and VH3 B01 express at ~1 g l−1 in shake flask culture) and have good stabilities (melting temperature (Tm) = 56–65 °C) (Supplementary Fig. 7). The most potent binders elute as a single monodisperse peak via SEC (Supplementary Fig. 8), and VH3 B01 retains binding to Spike-RBD and a monodisperse SEC profile after lyophilization and reconstitution, suggesting it could be suitable for lyophilized formulation (Supplementary Fig. 9).
Bi-paratopic and multivalent VH neutralize virus
SARS-CoV2 is a biosafety level 3 (BSL3) pathogen. To facilitate studies under routine laboratory conditions, we utilized a pseudotyped lentiviral (HIV) particle that has been reported previously36. These pseudotyped particles were generated from a three-plasmid system containing non-Env proteins from HIV, a luciferase reporter and the SARS-CoV-2 Spike protein. Entry of this viral particle into ACE2-expressing target cells and neutralization by anti-Spike antibodies have previously been shown to faithfully recapitulate features of the authentic pathogen without the need for working under BSL3 conditions. This pseudotyped virus was used to determine the half-maximal inhibitory concentration (IC50) of neutralization for each construct.
The VH monomers neutralized pseudotyped virus weakly (IC50 > 50 nM), and cocktails of unlinked monomers did not improve potency. By contrast, the multivalent binders (VH2, VH3 and VH-Fc) neutralized ~10–1,000-fold more potently than their respective monomeric units (Fig. 4a, Supplementary Table 5 and Extended Data Fig. 5). There was a linear correlation between the in vitro binding affinity (Kd) to Spike-RBD and the pseudotyped neutralization potency (IC50) across the different binders (R2 = 0.72) (Fig. 4b).

a, Pseudotyped virus IC50 values of VH binders. The neutralization potency improves when VH domains are engineered into multivalent and bi-paratopic constructs. b, Correlation of in vitro binding affinity (Kd) and pseudotyped virus neutralization (IC50) of VH binders. Data were fit to a log–log linear extrapolation. c, Pseudotyped virus neutralization curves of multi-site VH2 in comparison to single-site VH2 demonstrate that the multi-site VH2 have a more cooperative neutralization curve. d, Pseudotyped virus neutralization curves of mono-, bi- and trivalent formats of VH B01, demonstrating the potency gains driven by valency. e, Authentic SARS-CoV-2 neutralization curves for the most potent VH formats were determined via quantitative polymerase chain reaction (qPCR) of the viral genome in cellular RNA. All pseudoviral neutralization data were repeated as n = 2 independent replicates. Authentic virus neutralization data were repeated as n = 2 independent replicates. Data represent the average and standard deviation of replicates.
In particular, we observed that bi-paratopic VH2 A01-B01 and VH2 A01-B02 were stronger neutralizers than unlinked monomer cocktails. Additionally, the neutralization curves of the bi-paratopic (multi-site) VH2 differed from the homodimeric (single-site) VH2 binding to either site A or B. That is, the bi-paratopic VH2 exhibited a more cooperative transition and fully neutralized virus, while the homodimeric VH2 showed a more linear transition and did not fully block viral entry, even at high concentrations (Fig. 4c). This may reflect mechanistic differences—the bi-paratopic VH2 can theoretically engage a single RBD using both VH domains simultaneously (intra-RBD avidity) and more fully occlude the ACE2 binding site, while the homodimeric VH2 must bridge separate RBDs within the trimer (inter-RBD avidity).
Furthermore, the increase in neutralization potency as we increase the number of tandem VH units is exemplified by the VH B01-derived binders, as the IC50 values of VH2 B01 and VH3 B01 are two to three orders of magnitude lower than the IC50 of the VH B01 monomer (Fig. 4d). This is also observed for VH-Fc B01, which also neutralizes two orders of magnitude more potently than the monomer. Interestingly, although the neutralization potency of VH2 A01 is better than VH A01, the potency does not improve further when a third domain is added (VH3 A01), indicating that epitope-specific geometries can affect the extent to which increasing valency improves potency. The pseudotyped virus neutralization assays demonstrate that the top predicted binders from in vitro affinity data, VH2 A01-B01, VH2 A01-B02, VH3 B01 and VH-Fc B01, are indeed the most potent, with IC50 values of 0.74 nM, 1.08 nM, 0.156 nM and 1.86 nM, respectively.
Finally, we tested the ability of the most potent VH binders to neutralize authentic SARS-CoV-2 virus. As predicted, VH3-B01 neutralized most potently. The VH-Fc B01, VH2 A01-B01 and VH2 A01-B02 followed trends consistent with both the in vitro binding Kd and pseudotyped virus IC50. VH3 B01, VH-Fc B01, VH2 A01-B01 and VH2 A01-B02 blocked authentic SARS-CoV-2 viral entry with IC50 values of 3.98 nM, 33.5 nM, 12.0 nM and 26.2 nM, respectively (Fig. 4e and Supplementary Table 6).
Cryo-EM confirms multivalent binding of VH3 B01 to Spike
To confirm whether our linking strategy could successfully engage multiple RBDs on Spike, we obtained a 3.2-Å global resolution cryo-EM three-dimensional (3D) reconstruction of SARS-CoV-2 Secto in complex with VH3 B01, the most potent neutralizer (Fig. 5a, Extended Data Fig. 6, Supplementary Fig. 10 and Supplementary Table 7). Although the S2 region of Secto was resolved at the reported resolution, the RBDs with the bound VH domains were resolved at ~6-Å resolution. However, even at this resolution, the structure unambiguously showed that the three RBDs on Spike are in a two ‘up’ and one ‘down’ conformation. Densities corresponding to each VH domain are present on all three RBDs, indicating that VH B01 can bind both ‘up’ and ‘down’ conformations of RBD. SARS-CoV-2 Spike is rarely observed in this conformation, with most structures being in all RBD ‘down’ state or one RBD ‘up’ state4,6,32,37. The binding epitope of VH B01 overlaps with the known ACE2 binding site (Fig. 5b), confirming the intended mechanism of neutralization and validating the ability of the masked selection strategy to precisely direct a binder toward the intended surface on a target protein. To further investigate the dependence of binding on Spike conformation, we performed BLI experiments with Secto at pH 4.5, which has been reported recently to lock Spike in an ‘all-down’ RBD conformation38. Binding of both VH3 B01 and VH2 A01-B01 to Secto were greatly diminished (Extended Data Fig. 7). This observation suggests that one or more RBDs in the ‘up’ conformation may be required for VH B01 to access multiple RBDs on Spike.

a, Side and top views of cryo-EM 3D reconstructions of VH3 B01 + Secto (PDB 7JWB), shown with individual VH domain densities of VH3 B01 fit with PDB 3P9W (VH scaffold; orange cartoon). A total of three VH domains, each bound to an RBD of the Spike trimer, are resolved. The 3D model of Secto was fit with a reference structure (PDB 6X2B, with additional rigid body fit of the individual RBDs; blue cartoon) and shows RBDs in a distinct two ‘up’, one ‘down’ conformation. The cryo-EM map was low-pass-filtered to 6 Å. b, View of the epitope (site B) of one VH domain from VH3 B01. Site B overlays directly with the ACE2 binding site (yellow surface; contacts defined as RBD residues within 8 Å of an ACE2 residue from PDB 6M0J).
Discussion
Here, we describe a straightforward strategy to rapidly generate linked single-domain binders that potently neutralize SARS-CoV-2. We began by creating and validating a diverse human VH-phage library and generating VH binders to the ACE2 binding interface of Spike-RBD by a masked selection approach. From a panel of 85 unique VH binders, three were identified that recognized two separate epitopes within the ACE2 binding interface with nanomolar affinity. To engage multiple epitopes simultaneously within an RBD or across RBDs on Spike, these VH monomers were linked into multivalent and bi-paratopic formats by Gly-Ser linkers or Fc domains, without any further high-resolution structural information. This linkage approach not only significantly enhanced affinity, but also substantially improved the viral neutralization potency. We confirmed the basis of this increase by obtaining a cryo-EM structure of the most potent trivalent VH bound to Spike. Consistent with our original design, the trivalent VH simultaneously blocked the ACE2 binding site on all three RBDs of Spike.
We show that the in vitro binding affinities and neutralization potencies against this oligomeric target can be dramatically increased with valency. This is exemplified by VH B01, which shows a 460-fold increase in binding affinity and 1,400-fold increase in pseudotyped virus neutralization from VH B01 to VH2 B01 to VH3 B01. The cryo-EM structure of VH3 B01 shows that this binder neutralizes SARS-CoV-2 by blocking the ACE2 binding site on all three RBDs on Spike. Although there could be other contributing neutralization mechanisms such as viral aggregation induced by inter-Spike or inter-virion binding by the multivalent VH, the relatively short linker length (20 amino acids) between the VH domains would make these long-distance interactions significantly less likely. Additionally, no aggregates or Secto multimers induced by VH3 B01 were observed during cryo-EM sample preparation and data collection, which further suggests that inter-Spike or inter-virion binding effects, if present, are minimal.
A similar relationship between valency and potency is observed for VH B02, which also targets the same epitope (site B). However, VH A01, which targets site A, is mechanistically distinct, as there is no change in potency between a bivalent (VH2) and a trivalent (VH3) format. This suggests that, unlike site B binders, the trivalent site A binder may not be able to fully engage all three RBDs. This could be due to the specific binding mechanism and epitope of VH A01, conformational differences of the RBDs within the Spike trimer or spatial constraints of the linker. Structure determination of site A binders in complex with Spike can elucidate the mechanistic and geometric difference between site A and site B epitopes. Additionally, structure-guided approaches to optimize linker lengths and orientations, coupled with an affinity maturation campaign, may enable further increases in potency beyond what is demonstrated in this study.
Interestingly, we observed a difference in the cooperativity of the pseudotyped virus neutralization curves between bivalent VH2 that target both site A and site B (bi-paratopic) versus VH2 that target only site A or site B (mono-paratopic). Despite similar IC50 values, the IC95 values for bi-paratopic VH2 (VH2 A01-B01 and VH2 A01-B02) were much lower than mono-paratopic VH2 binders. This could indicate a mechanistic difference between these two types of bivalent binder. Bivalent VH2 can engage multiple RBDs, but only two of the three RBDs can be occluded efficiently, leaving one RBD accessible to ACE2. This could underlie why the neutralization of mono-paratopic VH2 does not reach 100% compared to, say, a trivalent VH3. Curiously, however, the bi-paratopic VH2 can fully neutralize virus, despite also having only two VH domains. We reason that intra-RBD avidity may play an important and unique role in the neutralization mechanism of bi-paratopic VH2. Mono-paratopic binders are limited to inter-RBD avidity and, regardless of valency, are limited to up to three binding sites on Spike. By contrast, bi-paratopic VH2 can utilize up to six binding epitopes on Spike and utilize both inter-RBD and intra-RBD avidity. Additionally, a more complete occlusion of the ACE2 binding interface (~864 Å2)39 on RBD by the bi-paratopic VH2 could also underlie this difference in neutralization profiles. Although we do not have a structure of VH2 A01-B01, we know both A01 and B01 can bind simultaneously within the ACE2 binding site. The cryo-EM structure for B01 shows good coverage of the ACE2 binding site (Fig. 5b) while leaving open adjacent space for VH A01 to occupy a non-overlapping epitope. A better understanding of the binding and neutralization mechanisms of bi-paratopic binders remain a current area of investigation and could lead to the engineering of more potent, multi-specific binders.
Previous studies that utilized designed ankyrin repeat proteins (DARPins), llama-derived nanobodies, computationally designed proteins and bivalent Fabs inspired our engineering strategy40,41,42,43,44,45,46. We believe our human-derived VH domains offer distinct advantages. Foremost, they would not require the time-consuming structure-guided humanization process that would be necessary for therapeutic nanobody development. Additionally, the VH domains derived from our library have favorable biophysical properties derived from a shared well-behaved scaffold. Our most potent construct, VH3 B01, retains high-affinity binding after lyophilization cycles. This factor, coupled with scalable production in bacterial systems, may enable lung delivery via inhalation and could facilitate rapid deployment in response to a pandemic. Despite these advantages, single-domain binders have comparatively lower stability and in vivo half-life than mAbs and, due to their novelty, pharmacodynamic properties, such as the prevalence of anti-drug antibodies, have not been as extensively tested in the clinic. Despite these challenges, the engineering potential of these single-domain binders is immense and provides an exciting avenue for the next generation of therapeutic antibody engineering and development. Our results illustrate this potential through a straightforward and rapid strategy to improve the efficacy of single-domain binders and could be applied to other protein interfaces of interest, including future viral proteins and other antigens associated with human disease.
Methods
VH library construction and validation by next-generation sequencing
The VH-phage library was created through bivalent display of VH on the surface of M13 bacteriophage, as previously described29,47,48. In brief, the DNA phagemid library was created through oligonucleotide mutagenesis. First, the human VH-4D5 sequence was modified with five mutations (35G/39R/45E/47L/50R) in the framework and with restriction sites in each of the CDRs: AgeI in CDR H1, NcoI in CDR H2 and XhoI in CDR H3 (Supplementary Table 1). Oligonucleotides were synthesized by a custom Trimer Phosphoramidite mix for CDR H1 and CDR H2 (Twist Bioscience) and CDR H3 (Trilink Biotechnologies) (Supplementary Table 2). After mutagenesis, DNA sub-library pools were digested with appropriate restriction enzymes to remove the phagemid template before transformation into SS320 electrocompetent cells (Lucigen) for phage production. NGS of the CDR H3 was performed on the pooled library by amplifying the phagemid from boiled phage with in-house primers. Samples were submitted for analysis on a HiSeq4000 (Illumina) system with a custom primer: TGAGGACACTGCCGTCTATTATTGTGCTCGC (Tm = 67 °C, GC content = 52%). NGS analysis of the output was performed using an in-house informatics pipeline written in R. In brief, the raw NGS data sequencing file (*.fastq.gz) was converted into a table composed of the DNA sequences, the amino-acid sequences (CDR H3) and the counts/frequency as columns, then saved as a *.csv file for further analysis (for example, calculation of amino acid abundancy, sequence logo, H3 length distribution and so on). Several filters were applied: (1) low-quality sequences containing ‘N’ were removed; (2) sequences with any stop codon were removed; (3) only the sequences that were in-frame were kept. Scripts are available for download at https://github.com/crystaljie/VH_library_CDR_H3_NGS_analysis_Cole.Bracken.git.
Cloning, protein expression and purification
Spike-RBD monomer, Spike-RBD-Fc, Spike ectodomain (Secto) and ACE2-Fc were produced as biotinylated proteins as previously described33. VH were subcloned from the VH-phagemid into E. coli expression vector pBL347. VH2 and VH3 were cloned into pBL347 with a 20-amino-acid Gly-Ser linker. VH-Fc were cloned into a pFUSE (InvivoGen) vector with a human IgG1 Fc domain. All constructs were sequence-verified by Sanger sequencing. VH, VH2 and VH3 constructs were expressed in E. coli C43(DE3) Pro+ using optimized autoinduction medium and purified by protein A affinity chromatography, similarly to Fabs (Supplementary Fig. 11)29. VH-Fc were expressed in Expi293 BirA cells using transient transfection (Expifectamine, Thermo Fisher Scientific). Four days after transfection, the medium was collected and VH-Fc were purified using protein A affinity chromatography. All proteins were buffer-exchanged into PBS by spin concentration and stored in aliquots at −80 °C. The purity and integrity of proteins were assessed by SDS-PAGE. All proteins were endotoxin-removed using an endotoxin removal kit (Thermo Fisher) before use in neutralization assays.
Phage selection with the VH-phage library
Phage selections were performed according to previously established protocols29, using biotinylated antigens captured with streptavidin-coated magnetic beads (Promega). In each round, the phage pool was first cleared by incubation with beads loaded with 500 nM ACE2-Fc/Spike-RBD-Fc complex. The unbound phage were then incubated with beads loaded with Spike-RBD-Fc. After washing, the bound phage was eluted by the addition of 2 μg ml−1 of tobacco etch virus protease. In total, four rounds of selection were performed with decreasing amounts of Spike-RBD-Fc, as shown in Fig. 2a. All steps were carried out in PBS buffer + 0.02% Tween-20 + 0.2% BSA (PBSTB). Individual phage clones from the third and fourth rounds of selections were analyzed by phage ELISA.
Phage enzyme-linked immunosorbent assay
For each phage clone, four different conditions were tested—direct: Spike-RBD-Fc; competition: Spike-RBD-Fc with an equal concentration of Spike-RBD-Fc in solution; negative selection: ACE2-Fc/Spike-RBD-Fc complex; control: Fc. Flat-bottomed clear plates (Nunc MaxiSorp 384-well, Thermo Fisher Scientific) were coated with 0.5 μg ml−1 of NeutrAvidin in PBS overnight at 4 °C and subsequently blocked with PBS + 0.02% Tween-20 + 2% BSA for 1 h at room temperature. Plates were washed three times with PBS containing 0.05% Tween-20 (PBST) and were washed similarly between each of the steps, then 20 nM biotinylated Spike-RBD-Fc, ACE2-Fc/Spike-RBD-Fc complex or Fc diluted in PBSTB was captured on the NeutrAvidin-coated wells for 30 min, then blocked with PBSTB + 10 μM biotin for 30 min. Phage supernatant diluted 1:5 in PBSTB was added for 20 min. For the competition samples, the phage supernatant was diluted into PBSTB with 20 nM Spike-RBD-Fc. Bound phage were detected by incubation with anti-M13 antibody conjugated to horseradish-peroxidase (HRP) (Sino Biological; 1:5,000) for 30 min, followed by the addition of 3, 3', 5, 5' tetramethyl benzidine (TMB) substrate (VWR International). The reaction was quenched with the addition of 1 M phosphoric acid, then absorbance at 450 nm was measured using a Tecan M200 Pro spectrophotometer.
Enzyme-linked immunosorbent assay half-maximum effective concentration with Spike-RBD-monomer
Flat-bottomed clear plates (384-well Nunc Maxisorp) were prepared similarly to the phage ELISA protocol (above) by coating with neutravidin, followed by blocking with PBST + 2% BSA, incubation with 20 nM Spike-RBD-monomer and blocking by PBSTB + 10 μM biotin. VH binders in fourfold dilutions ranging from 500 nM to 2.8 pM were added for 1 h. Bound VH was detected by incubation with Protein A HRP conjugate (Thermo Fisher Scientific; 1:10,000) for 30 min, followed by the addition of TMB substrate for 5 min, quenching by 1 M phosphoric acid and detection of absorbance at 450 nm. Each concentration was tested in duplicate and the assay repeated three times.
Bio-layer interferometry experiments
BLI data were measured using an Octet RED384 (ForteBio) instrument. Spike-RBD or Secto was immobilized on a streptavidin or Ni-NTA biosensor and loaded until a 0.4-nm signal was achieved. After blocking with 10 μM biotin, purified binders in solution were used as the analyte. PBSTB was used for all buffers for BLI at pH 7.4. For BLI at pH 4.5, 10 mM sodium acetate pH 4.5, 150 mM NaCl, 0.2 mg ml−1 BSA and 0.01% (wt/vol) Tween-20 were used. Data were analyzed using ForteBio Octet analysis software and kinetic parameters were determined using a 1:1 monovalent binding model.
Competition enzyme-linked immunosorbent assay with COVID-19 convalescent patient sera
Competition ELISA with convalescent patient sera was conducted with the same patient sera as previously reported35. Samples were collected in accordance with the Declaration of Helsinki using protocols approved by the UCSF Institutional Review Board (protocol 20-30338). Patients were voluntarily recruited based on their history of previous SARS-CoV-2 infection. All patients provided written consent. Patient sera were de-identified before delivery to the Wells Lab, where all experiments presented here were performed. Briefly, sera were obtained, as described, from patients with a history of a positive test with nasopharyngeal SARS-CoV-2 PCR with reverse transcription PCR (RT-PCR) and at least 14 days after the resolution of their COVID-19 symptoms. Healthy control serum was obtained before the emergence of SARS-CoV-2. Sera were heat-inactivated (56 °C for 60 min) before use. Competitive serology using biotinylated SARS-CoV-2 Spike-RBD as the capture antigen was performed as previously reported35, but with slight modifications. Instead of supplementing sera diluted 1:50 in 1% nonfat milk with 100 nM ACE2-Fc, each of the indicated VH-Fc fusions was used at 100 nM. Bound patient antibodies were then detected using Protein L-HRP (Thermo Fisher Scientific 32420, 1:5,000). Background from the raw ELISA signal in serum-treated wells was removed by first subtracting the signal measured in NeutrAvidin-alone coated wells, then subtracting the signal detected in antigen-coated wells incubated with 1% nonfat milk + 100 nM competitor.
Differential scanning fluorimetry
DSF was conducted as previously described29. Briefly, purified protein was diluted to 0.5 μM or 0.25 μM in buffer containing Sypro Orange 4x (Invitrogen) and PBS and assayed in a 384-well white PCR plate. All samples were tested in duplicate. In a Roche LC480 LightCycler, the sample was heated from 30 °C to 95 °C with a ramp rate of 0.3 °C per 30 s, and the fluorescence signals at 490 nm and 575 nm were continuously collected. Tm was calculated using the Roche LC480 LightCycler software.
Size exclusion chromatography
SEC analysis was performed using an Äkta Pure system (GE Healthcare) using a Superdex 200 Increase 10/300 GL column. A 100 μl volume of 2–3 mg ml−1 of each analyte was injected and run with a constant mobile phase of degassed 10 mM Tris pH 8.0 and 200 mM NaCl. The absorbance at 280 nm was measured. Post-lyophilization and reconstitution SEC was performed using an Agilent HPLC 1260 Infinity II LC system using an AdvanceBio SEC column (300 Å, 2.7 μm, Agilent). Fluorescence (excitation 285 nm, emission 340 nm) was measured.
Preparation of SARS-CoV-2 pseudotyped virus and the HEK-ACE2 overexpression cell line
HEK293T-ACE2 cells were a gift from A. Wiita’s laboratory at the University of California, San Francisco. Cells were cultured in D10 medium (DMEM + 1% pen-strep + 10% heat-inactivated FBS). Plasmids to generate pseudotyped virus were a gift from P. Kim’s lab at Stanford University and SARS-Cov-2 pseudotyped virus was prepared as previously described36. Briefly, plasmids at the designated concentrations were added to OptiMEM medium with FuGENE HD transfection reagent (Promega) at a 3:1 FuGENE:DNA ratio, incubated for 30 min, and subsequently transfected into HEK293T cells. After 24 h, the supernatant was removed and replaced with D10 culture medium. Virus was propagated for an additional 48 h, and the supernatant was collected and filtered. Virus was stored at 4 °C for up to 10 days.
HEK-ACE2 were seeded at 10,000 cells per well on 96-well white plates (Corning, cat. no. 354620). After 24 h, pseudotyped virus stocks were titered via a twofold dilution series in D10 medium, and 40 μl was added to cells. After 60 h, infection and the intracellular luciferase signal were determined using a Bright-Glo luciferase assay (Promega), and the dilution achieving maximal luminescent signal within the linear range, ~3–5 × 105 luminescence units, was chosen as the working concentration for neutralization assays. Pseudovirus stocks were flash-frozen in aliquots and stored at −80 °C and thawed on ice just before use in a neutralization assay.
Pseudotyped viral neutralization assays
HEK-ACE2 cells were seeded at 10,000 cells per well in 40 μl of D10 on 96-well white plates (Corning, cat. no. 354620) 24 h before infection. To determine the IC50 values for pseudotyped virus, dose series of each VH binder were prepared at 3× concentration in D10 medium and 50 μl samples were aliquoted into each well in a 96-well plate format. Next, 50 μl of virus diluted in D10 medium was added to each well and the virus and blocker solution were allowed to incubate for 1 h at 37 °C. Subsequently, 80 μl of the virus and blocker solution were transferred to wells seeded with HEK-ACE2. After 60 h of infection at 37 °C, the intracellular luciferase signal was measured using a Bright-Glo luciferase assay: 80 μl of reconstituted Bright-Glo luciferase reagent was added to each well, incubated at room temperature with gentle shaking for 5 min, before the luminescence was measured on a Tecan M200 Pro spectrophotometer. The IC50 was determined using a four-parameter nonlinear regression (GraphPad Prism).
Authentic viral neutralization assays
Authentic virus neutralization assays were carried out as previously described33. All handling and experiments using live SARS-CoV-2 virus clinical isolate 2019-nCoV/USA-WA1/2020 (BEI Resources) was conducted under BSL3 containment with approved Biohazard Use Authorization (BUA) and protocols. VeroE6 cells were cultured in minimal essential medium (MEM), 10% FBS and 1% pen-strep. For neutralization assays, VeroE6 cells were seeded on six-well culture plates at 3.8 × 105 cells per well the day before. Infection with SARS-CoV-2 was performed using a multiplicity of infection (MOI) of 0.1. Virus was incubated in infection medium (EMEM 0% FBS) containing different concentrations of binders for 1 h at 37 °C. Culture medium was removed from the VeroE6 cells and 300 μl of the blocker/virus inoculum was added to the cells for 1 h at 37 °C. After this step, 1 ml of EMEM with 10% FBS was added to the cells, and the cells were incubated at 37 °C for an additional 16 h before RNA collection. Viral entry into cells and cellular transcription of viral genes was measured by qPCR using the N gene and host beta-glucuronidase (GUSB) and host beta-actin (ACTB) as controls. An RNAeasy RNA extraction kit (Qiagen) was used for RNA extraction and a Quantitect reverse-transcriptase kit (Qiagen) was used to generate cDNA. qPCR reactions were prepared using SYBR Select Master Mix (Thermo). The N gene and hGUSB gene primer concentration was 400 nM and the annealing temperature was 58 °C. Primer sequences (IDT) were as follows—viral N gene: N_F = CACATTGGCACCCGCAATC; N_R = GAGGAACGAGAAGAGGCTTG; host gene: hGUSB_F = CTCATCTGGAATTTTGCCGATT; hGUSB_R = CCGAGTGAAGACCCCCTTTTTA. The relative copy number (RCN) of viral transcript level compared to the host transcript was determined using the ∆∆CT method. IC50 values were determined using a four-parameter nonlinear regression (GraphPad Prism).
Expression and purification of the Spike ectodomain for cryo-electron microscopy
To obtain the pre-fusion Spike ectodomain, methods similar to those in previous reports were used32,49. The expression plasmid, provided by the McLellan lab, was used in a transient transfection with 100 ml, high-density Chinese hamster ovary (ExpiCHO, Thermo Fisher) culture following the ‘High Titer’ protocol provided by Thermo Fisher. Six to nine days post transfection, the supernatant was collected with centrifugation at 4,000g at room temperature. The clarified supernatant was then incubated with Ni-Sepharose Excel resin (Cytiva Life Sciences) for 90 min at room temperature. After incubation, the nickel resin was washed with 20 mM Tris (pH 8), 200 mM NaCl and 20 mM imidazole with 10 column volumes. Protein was eluted from the nickel resin with 20 mM Tris (pH 8), 200 mM NaCl and 500 mM imidazole. Eluate was then concentrated with a 50 molecular weight cutoff Amicon Ultra-15 centrifugal unit by centrifugation at 2,500g at room temperature. The eluate was concentrated, filtered with a 0.2-µm filter and injected onto a Superose 6 10/300 GL column equilibrated with 10 mM Tris (pH 8) and 200 mM NaCl. Fractions corresponding to monodisperse Spike were collected and the concentration was determined using a NanoDrop system.
Cryo-electron microscopy sample preparation, data collection and processing
Spike ectodomain (2 µM) was mixed with fivefold excess VH3 B01 and applied (3 µl) to holey-carbon Au 200 mesh 1.2/1.3 Quantifoil grids. Grids were blotted and plunge-frozen using a Mark 4 Vitrobot (Thermo Fisher) at 4 °C and 100% humidity, utilizing blot force 0 and a blot time of 4 s. A total of 1,656 images were collected on a Titan Krios cryo-electron microscope (Thermo Fisher) equipped with a K3 direct detector operated in correlated double sampling (CDS) mode (Gatan Ametek) and an energy filter (Gatan Ametek) at a nominal magnification of ×105,000 (0.834 Å per physical pixel). Dose-fractionated videos were collected with a total exposure of 6 s and 0.04 s per frame at a dose rate of nine electrons per physical pixel per second. Videos were corrected for motion and filtered to account for electron damage utilizing MotionCor250. Drift-corrected sums were imported into the cryoSPARC2 processing package51. Micrographs were manually curated, contrast transfer function (CTF) was estimated utilizing patches, and particles were picked with a Gaussian blob. The previous Spike ectodomain structure was imported as an initial model (low-pass-filtered to 30 Å) and multiple rounds of 3D and 2D classification were performed. Images were re-picked with the best looking 2D class averages low-pass-filtered to 30 Å, and multiple rounds of 3D classification were performed again to obtain a homogeneous stack of Spike trimer particles. The majority of the particles went into classes putatively representing excess unbound VH3 B01 and the final Spike-like particle stack only contained ~21,000 particles. Non-uniform homogeneous refinement of the particle stack resulted in a global resolution of 3.2 Å (masked) utilizing 0.143 fourier shell correlation (FSC) cutoff52. PDB 6X2B was rigid body fit into the resulting reconstruction in UCSF Chimera53. The RBDs of two Spike trimers were moved as a rigid body to accommodate the cryo-EM density. The cryo-EM reconstruction was low-pass-filtered to 6 Å to better visualize the VH densities. The homology model was built based on PDB 4G80 for the VH domains, and the resulting and individual VH domains were rigid body fit into the 6-Å cryo-EM density as depicted in Fig. 5a. The resulting model was relaxed into the cryo-EM map low-pass-filtered to 6 Å with the Rosetta FastRelax protocol. For Supplementary Fig. 13 the local resolution was estimated using ResMap54. The final figures were prepared using ChimeraX55.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this Article.
Data availability
Cryo-EM structural data have been deposited in the Protein Data Bank and Electron Microscopy Data Bank, (PDB 7JWB and EMD-22514). The cryo-EM structure of the Spike trimer from PDB 6VSB and ACE2 from PDB 6M17 and PDB 6M0J were used for distance estimates. The cryo-EM structure of Spike trimer from PDB 6X2B and the crystal structure of a VH domain from PDB 3P9W were used for cryo-EM data processing. Source data are provided with this paper. Any additional information is available upon request.
Code availability
Code used to analyze the NGS data has been deposited in GitHub and is available for download at https://github.com/crystaljie/VH_library_CDR_H3_NGS_analysis_Cole.Bracken.git.
References
Yan, R. et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448 (2020).
Abraham, J. Passive antibody therapy in COVID-19. Nat. Rev. Immunol. 20, 401–403 (2020).
Robbiani, D. F. et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020).
Pinto, D. et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295 (2020).
Rogers, T. F. et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 7520, eabc7520 (2020).
Cao, Y. et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84 (2020).
Shi, R. et al. A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature 584, 120–124 (2020).
Miersch, S. et al. Scalable high throughput selection from phage-displayed synthetic antibody libraries. J. Vis. Exp. 1–15 (2015); https://doi.org/10.3791/51492
Nguyen, V. K., Hamers, R., Wyns, L. & Muyldermans, S. Camel heavy-chain antibodies: diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 19, 921–930 (2000).
Holt, L. J., Herring, C., Jespers, L. S., Woolven, B. P. & Tomlinson, I. M. Domain antibodies: proteins for therapy. Trends Biotechnol. 21, 484–490 (2003).
Ingram, J. R., Schmidt, F. I. & Ploegh, H. L. Exploiting nanobodies’ singular traits. Annu. Rev. Immunol. 36, 695–715 (2018).
Hamer-Casterman, C. & Atarchouch, T. et al. Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 (1998).
De Genst, E. et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl Acad. Sci. USA 103, 4586–4591 (2006).
Arbabi-Ghahroudi, M., Tanha, J. & MacKenzie, R. Prokaryotic expression of antibodies. Cancer Metastasis Rev. 24, 501–519 (2005).
Zimmermann, I. et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 7, 1–32 (2018).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Saerens, D., Ghassabeh, G. H. & Muyldermans, S. Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 8, 600–608 (2008).
Nilvebrant, J., Tessier, P. & Sidhu, S. Engineered autonomous human variable domains. Curr. Pharm. Des. 22, 6527–6537 (2016).
Rouet, R., Dudgeon, K., Christie, M., Langley, D. & Christ, D. Fully human VH single domains that rival the stability and cleft recognition of camelid antibodies. J. Biol. Chem. 290, 11905–11917 (2015).
Sun, Z. et al. Potent neutralization of SARS-CoV-2 by human antibody heavy-chain variable domains isolated from a large library with a new stable scaffold. MAbs 12, 1778435 (2020).
Huo, J. et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 27, 846–854 (2020).
Walter, J. D. et al. Sybodies targeting the SARS-CoV-2 receptor-binding domain. Preprint at bioRxiv https://doi.org/10.1101/2020.04.16.045419 (2020).
Palomo, C. et al. Trivalency of a nanobody specific for the human respiratory syncytial virus fusion glycoprotein drastically enhances virus neutralization and impacts escape mutant selection. Antimicrob. Agents Chemother. 60, 6498–6509 (2016).
Carter, P. et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl Acad. Sci. USA 89, 4285–4289 (1992).
Harmsen, M. M. et al. Llama heavy-chain V regions consist of at least four distinct subfamilies revealing novel sequence features. Mol. Immunol. 37, 579–590 (2000).
Ma, X., Barthelemy, P. A., Rouge, L., Wiesmann, C. & Sidhu, S. S. Design of synthetic autonomous VH domain libraries and structural analysis of a VH domain bound to vascular endothelial growth factor. J. Mol. Biol. 425, 2247–2259 (2013).
Dudgeon, K. et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl Acad. Sci. USA 109, 10879–10884 (2012).
Birtalan, S. et al. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J. Mol. Biol. 377, 1518–1528 (2008).
Hornsby, M. et al. A high throughput platform for recombinant antibodies to folded proteins. Mol. Cell. Proteomics 14, 2833–2847 (2015).
Martinko, A. J. et al. Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins. eLife 7, e31098 (2018).
Kabat, E. A., Wu, T. T., Reid-Miller, M., Perry, H. & Gottesman, K. Sequences of Proteins of Immunological Interest (US Department of Health and Human Services, Public Health Service, National Institutes of Health, 1987); https://doi.org/10.1016/0003-2697(84)90805-4
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020).
Lui, I. et al. Trimeric SARS-CoV-2 Spike interacts with dimeric ACE2 with limited intra-Spike avidity. Preprint at bioRxiv https://doi.org/10.1101/2020.05.21.109157 (2020).
Neuman, B. W. et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 80, 7918–7928 (2006).
Byrnes, J. R. et al. Competitive SARS-CoV-2 serology reveals most antibodies targeting the Spike receptor-binding domain compete for ACE2 binding. mSphere 5, e00802–e00820 (2020).
Crawford, K. H. D. et al. Protocol and reagents for pseudotyping lentiviral particles with SARS-CoV-2 spike protein for neutralization assays. Viruses 12, 513 (2020).
Walls, A. C. et al. Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 180, 281–292 (2020).
Zhou, T. et al. A pH-dependent switch mediates conformational masking of SARS-CoV-2 spike. Preprint at bioRxiv https://doi.org/10.1101/2020.07.04.187989 (2020).
Lan, J. et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220 (2020).
Dong, J. et al. Development of multi-specific humanized llama antibodies blocking SARS-CoV-2/ACE2 interaction with high affinity and avidity. Emerg. Microbes Infect. 9, 1034–1036 (2020).
Binz, H. K., Amstutz, P. & Plückthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 23, 1257–1268 (2005).
Binz, H. K., Stumpp, M. T., Forrer, P., Amstutz, P. & Plückthun, A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332, 489–503 (2003).
Binz, H. K. et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin® drug candidate. MAbs 9, 1262–1269 (2017).
Conrath, K. E., Lauwereys, M., Wyns, L. & Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 276, 7346–7350 (2001).
Strauch, E. M. et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 35, 667–671 (2017).
Galimidi, R. P. et al. Intra-Spike crosslinking overcomes antibody evasion by HIV-1. Cell 160, 433–446 (2015).
Lee, C. V. et al. High-affinity human antibodies from phage-displayed synthetic Fab libraries with a single framework scaffold. J. Mol. Biol. 340, 1073–1093 (2004).
Chen, G. & Sidhu, S. S. Design and generation of synthetic antibody libraries for phage display. Methods Mol. Biol. 1131, 113–131 (2014).
Wang, B. et al. Bivalent binding of a fully human IgG to the SARS-CoV-2 spike proteins reveals mechanisms of potent neutralization. Preprint at bioRxiv https://doi.org/10.1101/2020.07.14.203414 (2020).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single particle cryo-EM reconstruction. Preprint at bioRxiv https://doi.org/10.1101/2019.12.15.877092 (2019).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Acknowledgements
We thank members of the Wells Lab, particularly those working on COVID-19 projects, for their efforts and contributions. Specifically, we would like to thank J. Gramespacher for purifying binders and M. Nix (UCSF) for guidance on pseudotyped virus neutralization assays. Additionally, we thank the entire QCRG for its rapid large-scale collaborative effort. Specifically, we thank C. Puchades and C. Azumaya for their efforts in the optimization of cryo-EM grid freezing and data collection, T. Owens for assistance in protein purification, D. Diwanji for expression of Secto and A. Manglik (UCSF) for advice on cryo-EM experiments and protein purification. We also thank the laboratory of P. Kim (Stanford University) for providing plasmids for pseudotyped virus production. Finally, we thank M. Wilson, C. Chiu and R. Loudermilk (UCSF), as well as the patients, for providing convalescent sera. J.A.W. is supported by generous grants from NCI (R35 GM122451-01); the Chan-Zuckerberg Biohub, UCSF Program for Breakthrough Biomedical Research (PBBR); Fast Grants from Emergent Ventures at the Mercatus Center, George Mason University (#2154); and funding from The Harrington Discovery Institute (GA33116). S.A.L. is a Merck Fellow of the Helen Hay Whitney Foundation. K.S. is a Fellow of the Helen Hay Whitney Foundation. X.X.Z. is a Merck Fellow of the Damon Runyon Cancer Research Foundation, DRG-2297-17. J.R.B. and J.Z. are supported by the National Institutes of Health National Cancer Institute (F32 5F32CA239417 (J.R.B.) and 5F32CA236151-02 (J.Z.)). B.S.Z. is supported by the National Institutes of Health National Cancer Institute (T32, HL007185). N.J.R. and I.L. are supported by the National Science Foundation (GRFP). D.P.N. was the Connie and Bob Lurie Fellow of the Damon Runyon Cancer Research Foundation (DRG-2204-14) and supported by a UCSF-PBBR Postdoctoral Independent Research Award, which is partially funded by the Sandler Foundation.
QCRG Structural Biology Consortium. In addition to those listed explicitly in the Author contributions, the structural biology portion of this work was performed by the QCRG (Quantitative Biosciences Institute Coronavirus Research Group) Structural Biology Consortium. Listed below are the contributing members of the consortium listed by teams in order of team relevance to the published work. The team leads (responsible for organization of each team and for the experimental design utilized within each team) are as follows: Cryo-EM grid freezing/collection team—Caleigh M. Azumaya, Cristina Puchades, Ming Sun; Cryo-EM data processing team—Miles Sasha Dickinson, Henry C. Nguyen; Mammalian cell expression team—Christian Billesboelle, Melody G. Campbell, Devan Diwanji, Carlos Nowotny, Amber M. Smith, Jianhua Zhao; Protein purification team—Daniel Asarnow, Michelle Moritz, Tristan W. Owens, Sergei Pourmal; Crystallography team—Nadia Herrera, Huong T. Kratochvil, Ursula Schulze-Gahmen, Iris D. Young; Bacterial expression team—Amy Diallo, Meghna Gupta, Erron W. Titus; Leadership team—Oren S. Rosenberg, Kliment A Verba. The QCRG Structural Biology Consortium has received support from the Quantitative Biosciences Institute, the Defense Advanced Research Projects Agency HR0011-19-2-0020 (to D.A.A. and K.A.V.; B. Shoichet PI), a FastGrants COVID19 grant (K.A. Verba PI), the Laboratory for Genomics Research (O.S. Rosenberg PI) and the Laboratory For Genomics Research (R.M. Stroud PI).
Author information
Authors and Affiliations
Consortia
Contributions
C.J.B. and D.P.N. designed and constructed the VH-phage library. C.J.B., S.A.L., N.J.R., J.Z., I.L., J.L. and K.P. cloned, expressed and purified the VH binders and/or antigens. C.J.B., S.A.L. and J.L. designed and/or conducted the in vitro characterization of the VH binders. J.R.B. designed and conducted the competition ELISA assay with convalescent patient sera. P.S., N.J.R. and S.A.L. designed and conducted the pseudovirus neutralization assays. P.S., N.J.R., B.S.Z. and S.A.L. designed and conducted the live virus neutralization assays. K.S. prepared samples for cryo-EM and the QCRG SBC performed cryo-EM data collection and analyses. S.A.L., X.X.Z., K.K.L. and J.A.W. led the coordination of external collaborations and supervised the project. C.J.B., S.A.L. and J.A.W. co-wrote the manuscript with input from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CDR loop design of VH-phage library.
Schematic of CDR amino acid composition as compared to parental template. Positions in pink highlight CDR H1 charged amino acid insertions. Positions in blue highlight the insertion of ‘X’ synthetic amino acid mixture. Positions in gray remain unchanged from template.
Extended Data Fig. 2 Specificity of lead VH domain binders.
Bio-layer interferometry (BLI) binding traces of VH domains at 150 nM to RBD and two decoy antigens expressed as biotinylated Fc-fusions: programmed cell death protein 1 (PD-1), and platelet glycoprotein 4 (CD36). Respective VH show successful binding to RBD, but not to either of the decoy antigens.
Extended Data Fig. 4 ELISA of VH binders to Spike-RBD.
ELISA of VH binders against Spike-RBD. Data presented show the average and standard deviation from three independent experiments. Data were fit to a non-linear, four-parameter variable slope regression model using Prism 8 to obtain EC50 values for each binder.
Extended Data Fig. 5 Pseudotyped virus neutralization by VH Binders.
Pseudotyped virus neutralization assays of VH binders. Data represent average and standard deviation of two biological replicates. Data were fit to a non-linear, four-parameter variable slope regression model using Prism 8 to obtain IC50 values.
Extended Data Fig. 6 Cryo-EM reconstruction of SARS-CoV-2 Spike trimer.
The SARS-CoV-2 Spike trimer + VH3 B01 cryo-EM reconstruction from non-uniform refinement in cryoSPARC at two different thresholds colored by resolution in the range from 3 Å to 10 Å. At high threshold the core S2 clearly displays high resolution features but the periphery of the molecule is closer to 6-7 Å.
Extended Data Fig. 7 pH dependent binding of lead multivalent and bi-paratopic VH.
BLI assay of lead constructs at 25 nM to Secto and RBD at pH 7.4 or pH 4.5. Binding of VH leads is only observed with Secto at pH 7.4 and RBD at pH 4.5.
Supplementary information
Supplementary Information
Supplementary Figs. 1–11 and Tables 1–7.
Source data
Source Data Fig. 1
NGS count source data.
Source Data Fig. 2
BLI source data.
Source Data Fig. 3
BLI, competition serology source data.
Source Data Fig. 4
Pseudoviral source data.
Source Data Extended Data Fig. 2
BLI source data.
Source Data Extended Data Fig. 4
ELISA source data.
Source Data Extended Data Fig. 5
Pseudoviral source data.
Source Data Extended Data Fig. 7
BLI source data.
Rights and permissions
About this article

Cite this article
Bracken, C.J., Lim, S.A., Solomon, P. et al. Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2. Nat Chem Biol 17, 113–121 (2021). https://doi.org/10.1038/s41589-020-00679-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-020-00679-1
This article is cited by
-
Computational design and engineering of self-assembling multivalent microproteins with therapeutic potential against SARS-CoV-2
Journal of Nanobiotechnology (2024)
-
Discovery of VH domains that allosterically inhibit ENPP1
Nature Chemical Biology (2024)
-
The role of single-domain antibodies (or nanobodies) in SARS-CoV-2 neutralization
Molecular Biology Reports (2022)
-
Passive Immunotherapy Against SARS-CoV-2: From Plasma-Based Therapy to Single Potent Antibodies in the Race to Stay Ahead of the Variants
BioDrugs (2022)
-
Arsenal of nanobodies shows broad-spectrum neutralization against SARS-CoV-2 variants of concern in vitro and in vivo in hamster models
Communications Biology (2022)
