
Abstract
Untargeted mass spectrometry is employed to detect small molecules in complex biospecimens, generating data that are difficult to interpret. We developed Qemistree, a data exploration strategy based on the hierarchical organization of molecular fingerprints predicted from fragmentation spectra. Qemistree allows mass spectrometry data to be represented in the context of sample metadata and chemical ontologies. By expressing molecular relationships as a tree, we can apply ecological tools that are designed to analyze and visualize the relatedness of DNA sequences to metabolomics data. Here we demonstrate the use of tree-guided data exploration tools to compare metabolomics samples across different experimental conditions such as chromatographic shifts. Additionally, we leverage a tree representation to visualize chemical diversity in a heterogeneous collection of samples. The Qemistree software pipeline is freely available to the microbiome and metabolomics communities in the form of a QIIME2 plugin, and a global natural products social molecular networking workflow.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The MS data, metadata and methods for the evaluation dataset have been deposited on the GNPS/MassIVE public repository2,32 under the accession number MSV000083306. Source data for the figures are available as Supplementary Datasets 2–5. The parameters used for molecular networking are available on GNPS at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=efda476c72724b29a91693a108fa5a9d. The chemical hierarchy generated by Qemistree (v.2020.1.2) is available on iTOL24 at https://itol.embl.de/tree/709513416494381587432576. The MS data, metadata and methods for Global FoodOmics dataset have been deposited on the GNPS/MassIVE public repository2,32 under the accession number MSV000085226. The parameters used for molecular networking are available on GNPS at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=ceb28a199d6b4f4fbf08490d9c96d631. The chemical hierarchy generated by Qemistree (v.2020.1.2) is available on iTOL24 at https://itol.embl.de/tree/13711034118313741584046018. The MS data, metadata and methods for Cheetah fecal dataset have been deposited on the GNPS/MassIVE public repository2,32 under the accession number MSV000082969. The parameters used for molecular networking are available on GNPS at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=093798dffe2448239410c3d465ef9fea.
Code availability
All source code is publicly available under BSD-2-Clause on GitHub at https://github.com/biocore/q2-qemistree. Qemistree is also available as an advanced analysis workflow on GNPS at https://ccms-ucsd.github.io/GNPSDocumentation/qemistree/. All analyses are documented in Jupyter Notebooks available at https://github.com/knightlab-analyses/qemistree-analyses.
References
Watrous, J. et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl Acad. Sci. USA 109, E1743–E1752 (2012).
Wang, M. et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 34, 828–837 (2016).
Fox Ramos, A. E., Evanno, L., Poupon, E., Champy, P. & Beniddir, M. A. Natural products targeting strategies involving molecular networking: different manners, one goal. Nat. Prod. Rep. 36, 960–980 (2019).
Böcker, S. & Dührkop, K. Fragmentation trees reloaded. J. Chem. Inform. 8, 5 (2016).
Rasche, F. et al. Identifying the unknowns by aligning fragmentation trees. Anal. Chem. 84, 3417–3426 (2012).
Washburne, A. D. et al. Phylogenetic factorization of compositional data yields lineage-level associations in microbiome datasets. PeerJ 5, e2969 (2017).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992).
Janssen, S. et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3, e00021–18 (2018).
McDonald, D. et al. Striped UniFrac: enabling microbiome analysis at unprecedented scale. Nat. Methods 15, 847–848 (2018).
Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 11, 1046–1053 (2006).
Heinonen, M., Shen, H., Zamboni, N. & Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 28, 2333–2341 (2012).
Laponogov, I., Sadawi, N., Galea, D., Mirnezami, R. & Veselkov, K. A. ChemDistiller: an engine for metabolite annotation in mass spectrometry. Bioinformatics 34, 2096–2102 (2018).
Dührkop, K., Shen, H., Meusel, M., Rousu, J. & Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl Acad. Sci. USA 112, 12580–12585 (2015).
Fan, Z., Ghaffari, K., Alley, A. & Ressom, H. W. Metabolite identification using artificial neural network. In Proc. 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 244–248 (IEEE, 2019).
Li, Y., Kuhn, M., Gavin, A.-C. & Bork, P. Identification of metabolites from tandem mass spectra with a machine learning approach utilizing structural features. Bioinformatics 36, 1213–1218 (2020).
Dührkop, K. et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019).
Pluskal, T., Castillo, S., Villar-Briones, A. & Oresic, M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 11, 395 (2010).
Nothias, L. et al. Feature-based molecular networking in the GNPS analysis environment.Nat. Methods 17, 905–908 (2020).
Treutler, H. et al. Discovering regulated metabolite families in untargeted metabolomics studies. Anal. Chem. 88, 8082–8090 (2016).
Depke, T., Franke, R. & Brönstrup, M. Clustering of MS2 spectra using unsupervised methods to aid the identification of secondary metabolites from Pseudomonas aeruginosa. J. Chromatogr. B 1071, 19–28 (2017).
Rawlinson, C. et al. Hierarchical clustering of MS/MS spectra from the firefly metabolome identifies new lucibufagin compounds. Sci. Rep. 10, 6043 (2020).
Schymanski, E. L. et al. Critical assessment of small molecule identification 2016: automated methods. J. Cheminform. 9, 22 (2017).
Feunang, Y. D. et al. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 8, 61 (2016).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Morton, J. T. et al. Learning representations of microbe-metabolite interactions. Nat. Methods 16, 1306–1314 (2019).
van der Hooft, J. J. J., Wandy, J., Barrett, M. P., Burgess, K. E. V. & Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl Acad. Sci. USA 113, 13738–13743 (2016).
Sedio, B. E., Rojas Echeverri, J. C., Boya, P. C. A. & Joseph Wright, S. Sources of variation in foliar secondary chemistry in a tropical forest tree community. Ecology 98, 616–623 (2017).
Bray, J. R., Roger Bray, J. & Curtis, J. T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Gauglitz, J. M. et al. Metabolome-informed microbiome analysis refines metadata classifications and reveals unexpected medication transfer in captive cheetahs. mSystems 5, e00635–19 (2018).
Thompson, L. R. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017).
Garland, T., Harvey, P. H. & Ives, A. R. Procedures for the analysis of comparative data using phylogenetically independent contrasts. Syst. Biol. 41, 18 (1992).
Junker, R. R. A biosynthetically informed distance measure to compare secondary metabolite profiles. Chemoecology 28, 29–37 (2017).
Bajusz, D., Rácz, A. & Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Chem. Inform. 7, 20 (2015).
Kuczynski, J. et al. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat. Methods 7, 813–819 (2010).
Gonzalez, A. et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798 (2018).
McDonald, D. et al. American Gut: an Open platform for citizen science microbiome research. mSystems 3, e00031-18 (2018).
Sinha, R., Abnet, C. C., White, O., Knight, R. & Huttenhower, C. The microbiome quality control project: baseline study design and future directions. Genome Biol. 16, 276 (2015).
Wang, M. et al. Assembling the community-scale discoverable human proteome. Cell Syst. 7, 412–421.e5 (2018).
Sumner, L. W. et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221 (2007).
Ludwig, M. et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2, 629–641 (2020).
Lozupone, C. A. & Knight, R. Species divergence and the measurement of microbial diversity. FEMS Microbiol. Rev. 32, 557–578 (2008).
Dührkop, K., Hufsky, F. & Böcker, S. Molecular formula identification using isotope pattern analysis and calculation of fragmentation trees. Mass Spectrom. 3, S0037 (2014).
Simón-Manso, Y. et al. Metabolite profiling of a NIST Standard Reference Material for human plasma (SRM 1950): GC-MS, LC-MS, NMR, and clinical laboratory analyses, libraries, and web-based resources. Anal. Chem. 85, 11725–11731 (2013).
Martens, L. et al. mzML-a community standard for mass spectrometry data. Mol. Cell. Proteom. 10, R110.000133 (2011).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Gauglitz, J. M. et al. Untargeted mass spectrometry-based metabolomics approach unveils molecular changes in raw and processed foods and beverages. Food Chem. 302, 125290 (2020).
Acknowledgements
P.C.D. was supported by the Gordon and Betty Moore Foundation (grant no. GBMF7622), CCF foundation no. 675191, the US National Institutes of Health (grant nos. U19 AG063744 01, P41 GM103484, R03 CA211211, R01 GM107550, 1 DP1 AT010885, P30 DK120515) and the University of Wisconsin-Madison OVCRGE; L.F.N. was supported by the US National Institutes of Health (grant no. R01 GM107550), and the European Union’s Horizon 2020 program (MSCA-GF, 704786). J.J.J.v.d.H. was supported by an ASDI eScience grant no. ASDI.2017.030, from the Netherlands eScience Center—NLeSC. K.D., M.F., M.L. and S.B. were supported by Deutsche Forschungsgemeinschaft (BO 1910/20). Y.V.B. was funded by the Janssen Human Microbiome Initiative through the Center for Microbiome Innovation at UC San Diego.
Author information
Authors and Affiliations
Contributions
A.T. and P.C.D. conceived the concept and managed the project. A.T. and Y.V.B. developed the algorithm and wrote the code for Qemistree. A.T. and Y.V.B. contributed equally to the work. L.F.N., R.K. and P.C.D. supervised method implementation. K.D., M.W., J.J.J.v.d.H., M.E., D.M. and A.G. tested and provided suggestions on how to improve the method. M.W. managed the deployment of Qemistree on GNPS. A.T. and M.W. developed the GNPS-Qemistree Dashboard. D.A. and A.T. wrote the documentation for the GNPS-Qemistree workflow. Y.V.B., Q.Z. and A.T. developed Qemistree-iTOL visualization. L.F.N. and M.N.E. performed the MS for the evaluation dataset. A.T., Y.V.B. and L.F.N. analyzed and interpreted the evaluation data. J.M.G. performed MS of the Global FoodOmics samples. A.T. and J.M.G. analyzed and interpreted the Global FoodOmics data. A.D.B. made the comparisons to CSCS. KD, M.F., M.L. and S.B. supported the integration of SIRIUS, ZODIAC and CSI:FingerID. A.T., Y.V.B., R.K. and P.C.D. wrote the manuscript. L.F.N., J.M.G., M.N.E., J.J.J.v.d.H., M.E., K.D., Q.Z., D.M., A.D.B., A.G., J.H., M.F., M.L. and S.B. improved the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.W. is a founder of Ometa Laboratories LLC. P.C.D. is a scientific advisor for Sirenas, Cybele and Galileo PCD is also a scientific advisor and founde of Enveda ans Ometa Laboratories LLC with approval by University of California San Diego. LLC. K.D., M.L., M.F. and S.B. are founders of Bright Giant GmbH.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
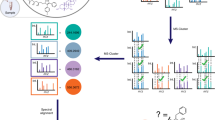
Extended Data Fig. 1 End-to-end Qemistree analysis using GNPS and QIIME2.
Qemistree analysis can be performed using two required input files: 1) A table of molecule (or chemical feature) abundances per sample and 2) an MGF file with MS1 and MS2 ion information. These inputs can be generated by processing mass spectrometry files (.mzXML) through MZmine for feature detection. In Qemistree, these input files are processed through SIRIUS and CSI:FingerID to generate molecular fingerprints and in silico structural annotations (SMILES) per MS feature. We use the predicted molecular fingerprints to generate a phenetic tree of relationships between MS features based on sub-structural similarity. This tree can be visualized in iTOL for further data exploration. If the user inputs a sample metadata file, they can also visualize the abundances of each MS feature stratified by sample grouping of interest. Additionally, the Qemistree queries ClassyFire to classify the structural annotations into chemical ‘kingdom’, ‘superclass’, ‘class’, ‘subclass’ and ‘direct parent’. We further allow the users to input a file with MS/MS spectral library matches (optional) into the workflow such that these library matches (typically, 2-20% of all MS features), instead of in silico annotation, are used for ClassyFire queries whenever available. All the outputs of the Qemistree workflow can be analyzed further using QIIME 2 tools (such as tree-based alpha and beta diversity, mmvec: https://github.com/biocore/mmvec, songbird: https://github.com/biocore/songbird) or explored in Python, R etc. as needed.
Extended Data Fig. 2
2D map of the LC-MS/MS data of the pooled sample for the C18 chromatographic conditions.
Extended Data Fig. 3
2D map of the LC-MS/MS data of the pooled sample for the C18-RTshift chromatographic conditions.
Extended Data Fig. 4 Technical variation in mass-spectrometry due to chromatographic shifts.
Sample (y-axis) by molecule (x-axis) heatmap of 2 fecal samples, tomato seedling samples, and serum samples in the evaluation dataset grouped by chromatography conditions.
Extended Data Fig. 5 Qemistree reduces the differences between biological replicates across mass-spectrometry runs.
A comparison of distances between sample replicates within and across chromatography gradients when using tree-agnostic (Bray-Curtis) distances and tree-based (Weighted UniFrac) distances.
Extended Data Fig. 6 Qemistree mitigates plate-to-plate variation in fecal metabolomics study to highlight a biologically-relevant effect.
a) Principal coordinate analysis (PCoA) of tree-agnostic distances (Bray-Curtis) colored by plate number (pseudo-F = 32.39, p = 0.001). b) PCoA of tree-informed distances (Weighted UniFrac) colored by plate number (pseudo-F = 15.67, p = 0.001). The same PCoA of (c) Bray-Curtis distances (pseudo-F = 33.50, p = 0.001) and (d) Weighted UniFrac distances (pseudo-F = 48.42, p = 0.001) colored by cheetah location which governed the diet of cheetahs. CBC: Cheetah Breeding Center; WD: Wildlife Discoveries.
Extended Data Fig. 7 Chemical taxonomy of food-derived compounds at chemical superclass level.
Chemical hierarchy of compounds (tree tips) detected in simple food products (single ingredient foods, N = 119). Internal nodes are labeled by pie charts of the superclass level taxonomy of children tips. Outer ring shows the relative abundance of each compound across simple animal products, plant products, and other (fungi and algae). The chemical hierarchy iTOL link: https://itol.embl.de/tree/7095134164128581587333337.
Extended Data Fig. 8 Chemical taxonomy of food-derived compounds at chemical class level.
Chemical hierarchy of compounds (tree tips) detected in simple food products (single ingredient foods, N = 119). Internal nodes are labeled by pie charts of the class level taxonomy of children tips. Outer ring shows the relative abundance of each compound across simple animal products, plant products, and other (fungi and algae). The chemical hierarchy iTOL link: https://itol.embl.de/tree/7095134164128581587333337.
Extended Data Fig. 9 Chemical hierarchy of the compounds observed in simple foods and seven complex samples.
a,b,c) 2 meals of orange chicken, a cooked cucumber and the sauce from a meal (schmorgurken), sour cream, blueberry kefir, and egg scramble with chorizo (N = 126 samples). The inner ring shows the relative abundance of each compound across simple animal products, plant products, fungi and algae (other) and complex foods. The absolute abundances of compounds in blueberry kefir (a), scrambled eggs with chorizo (b), and orange chicken (c) (outer bars) are overlaid on the tree to illustrate the shared and unique chemistry of complex foods. We highlight a classifier subtree annotated as benzodioxoles, compounds found in black pepper (in black) that are almost exclusively detected in complex foods. Note that untargeted mass-spectrometry is blind to stereochemistry and oftentimes regiochemistry (for example double bonds in a fatty acid); the structures shown are based on the spectral annotation of the reference library.
Supplementary information
Supplementary Information
Supplementary Tables 1–4.
Supplementary Dataset 1
Table of molecular properties (n = 2,936 properties) predicted by Sirius and CSI:FingerID.
Supplementary Dataset 2
Statistical source data for evaluation dataset (Figs. 1 and 2 and Extended Data Figs. 4 and 5).
Supplementary Dataset 3
Statistical source data for simple food samples (Fig. 3 and Extended Data Figs. 7 and 8).
Supplementary Dataset 4
Statistical source data for complex food samples (Fig. 4 and Extended Data Fig. 9).
Supplementary Dataset 5
Statistical source data for cheetah dataset (Extended Data Fig. 6).
Rights and permissions
About this article

Cite this article
Tripathi, A., Vázquez-Baeza, Y., Gauglitz, J.M. et al. Chemically informed analyses of metabolomics mass spectrometry data with Qemistree. Nat Chem Biol 17, 146–151 (2021). https://doi.org/10.1038/s41589-020-00677-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-020-00677-3
This article is cited by
-
Small molecule metabolites: discovery of biomarkers and therapeutic targets
Signal Transduction and Targeted Therapy (2023)
-
Annotating metabolite mass spectra with domain-inspired chemical formula transformers
Nature Machine Intelligence (2023)
-
Microbiomes and metabolomes of dominant coral reef primary producers illustrate a potential role for immunolipids in marine symbioses
Communications Biology (2023)
-
Recent advances in proteomics and metabolomics in plants
Molecular Horticulture (2022)
-
Salivary bacterial signatures in depression-obesity comorbidity are associated with neurotransmitters and neuroactive dipeptides
BMC Microbiology (2022)
