
Abstract
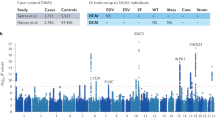
Brugada syndrome (BrS) is a cardiac arrhythmia disorder associated with sudden death in young adults. With the exception of SCN5A, encoding the cardiac sodium channel NaV1.5, susceptibility genes remain largely unknown. Here we performed a genome-wide association meta-analysis comprising 2,820 unrelated cases with BrS and 10,001 controls, and identified 21 association signals at 12 loci (10 new). Single nucleotide polymorphism (SNP)-heritability estimates indicate a strong polygenic influence. Polygenic risk score analyses based on the 21 susceptibility variants demonstrate varying cumulative contribution of common risk alleles among different patient subgroups, as well as genetic associations with cardiac electrical traits and disorders in the general population. The predominance of cardiac transcription factor loci indicates that transcriptional regulation is a key feature of BrS pathogenesis. Furthermore, functional studies conducted on MAPRE2, encoding the microtubule plus-end binding protein EB2, point to microtubule-related trafficking effects on NaV1.5 expression as a new underlying molecular mechanism. Taken together, these findings broaden our understanding of the genetic architecture of BrS and provide new insights into its molecular underpinnings.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
Data from the Genome Aggregation Database (gnomAD, v.2.1) are available at https://gnomad.broadinstitute.org. Data from the UK Biobank participants can be requested from the UK Biobank Access Management System (https://bbams.ndph.ox.ac.uk). Data from the Genotype Tissue Expression (GTEx) consortium are available at the GTEx portal (https://gtexportal.org) accessed December 2019 and March 2020. The Molecular Signatures Database (MSigDB, v.6.2) is available at http://www.gsea-msigdb.org/gsea/index.jsp. Other datasets generated during and/or analyzed during the current study can be made available upon reasonable request to the corresponding authors. Individual-level data sharing is subject to restrictions imposed by patient consent and local ethics review boards. The Brugada syndrome GWAS summary statistics are available on Zenodo, at https://doi.org/10.5281/zenodo.5095177 and on the GWAS catalog database (study ID accession: GCST90086158). The BrS polygenic score is available on the PGScatalog (PGS ID accession: PGS001779).
Change history
26 April 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41588-022-01079-y
References
Brugada, P. & Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396 (1992).
Priori, S. G. et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 36, 2793–2867 (2015).
Chen, Q. et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296 (1998).
Le Scouarnec, S. et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum. Mol. Genet. 24, 2757–2763 (2015).
Bezzina, C. R. et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 45, 1044–1049 (2013).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Mizusawa, Y. & Wilde, A. A. M. Brugada syndrome. Circ. Arrhythm. Electrophysiol. 5, 606–616 (2012).
van den Boogaard, M. et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852 (2014).
van Eif, V. W. W., Devalla, H. D., Boink, G. J. J. & Christoffels, V. M. Transcriptional regulation of the cardiac conduction system. Nat. Rev. Cardiol. 15, 617–630 (2018).
Gaborit, N. et al. Cooperative and antagonistic roles for Irx3 and Irx5 in cardiac morphogenesis and postnatal physiology. Development 139, 4007–4019 (2012).
Shen, T. et al. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. J. Clin. Invest. 121, 4640–4654 (2011).
Veerman, C. C. et al. The Brugada syndrome susceptibility gene HEY2 modulates cardiac transmural ion channel patterning and electrical heterogeneity. Circ. Res. 121, 537–548 (2017).
Tarradas, A. et al. Transcriptional regulation of the sodium channel gene (SCN5A) by GATA4 in human heart. J. Mol. Cell. Cardiol. 102, 74–82 (2017).
Arnolds, D. E. et al. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Invest. 122, 2509–2518 (2012).
Braz, J. C. et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10, 248–254 (2004).
Goldspink, D. A. et al. The microtubule end-binding protein EB2 is a central regulator of microtubule reorganisation in apico-basal epithelial differentiation. J. Cell. Sci. 126, 4000–4014 (2013).
Ajima, R. et al. Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrations. Genes Cells 13, 987–999 (2008).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
GTEx Consortium et al.Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Iotchkova, V. et al. GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat. Genet. 51, 343–353 (2019).
Gu, C. et al. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron 52, 803–816 (2006).
Wilde, A. A. M. et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell. Cardiol. 49, 543–553 (2010).
Talmud, P. J. et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 381, 1293–1301 (2013).
Lahrouchi, N. et al. Transethnic genome-wide association study provides insights in the genetic architecture and heritability of long QT syndrome. Circulation 142, 324–338 (2020).
Sotoodehnia, N. et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 42, 1068–1076 (2010).
van Setten, J. et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat. Commun. 9, 2904 (2018).
Arking, D. E. et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat. Genet. 46, 826–836 (2014).
Roselli, C. et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 50, 1225–1233 (2018).
Nielsen, J. B. et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 50, 1234–1239 (2018).
Rivaud, M. R. et al. A common co-morbidity modulates disease expression and treatment efficacy in inherited cardiac sodium channelopathy. Eur. Heart J. 39, 2898–2907 (2018).
Priori, S. G. et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 15, 1389–1406 (2013).
Al-Khatib, S. M. et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 138, e210–e271 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423 (2015).
Whiffin, N. et al. CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation. Genet. Med. 20, 1246–1254 (2018).
Kapplinger, J. D. et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46 (2010).
Walsh, R., Peters, N. S., Cook, S. A. & Ware, J. S. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J. Med. Genet. 51, 35–44 (2014).
Denham, N. C. et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 30, 118–127 (2019).
Walsh, R. et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of HCM. Genome Med. 11, 5 (2019).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Lee, S. H., Wray, N. R., Goddard, M. E. & Visscher, P. M. Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet. 88, 294–305 (2011).
Vutthikraivit, W. et al. Worldwide prevalence of Brugada syndrome: a systematic review and meta-analysis. Acta Cardiol. Sin. 34, 267–277 (2018).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Hormozdiari, F. et al. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 99, 1245–1260 (2016).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Schmitt, A. D. et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059 (2016).
Pers, T. H., Timshel, P. & Hirschhorn, J. N. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics 31, 418–420 (2015).
Leeuw, C. A., de, Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Backenroth, D. et al. FUN-LDA: a latent dirichlet allocation model for predicting tissue-specific functional effects of noncoding variation: methods and applications. Am. J. Hum. Genet. 102, 920–942 (2018).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Aragam, K. G. et al. Phenotypic refinement of heart failure in a national biobank facilitates genetic discovery. Circulation http://dx.doi.org/CIRCULATIONAHA.118.035774 (2018).
Choi, S. H. et al. Monogenic and polygenic contributions to atrial fibrillation risk: results from a national biobank. Circ. Res. 126, 200–209 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Panáková, D., Werdich, A. A. & Macrae, C. A. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature 466, 874–878 (2010).
Kuo, H.-H. et al. Negligible-cost and weekend-free chemically defined human iPSC culture. Stem Cell Rep. 14, 256–270 (2020).
Burridge, P. W., Holmström, A. & Wu, J. C. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr. Protoc. Hum. Genet. 87, 21.3.1–21.3.15 (2015).
Burridge, P. W. et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 11, 855–860 (2014).
Veerman, C. C. et al. Immaturity of human stem-cell-derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev. 24, 1035–1052 (2015).
Barry, P. H. & Lynch, J. W. Liquid junction potentials and small cell effects in patch-clamp analysis. J. Membr. Biol. 121, 101–117 (1991).
Bravo, E. et al. Developing a guideline to standardize the citation of bioresources in journal articles (CoBRA). BMC Med. 13, 33 (2015).
Wang, Y. et al. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Acknowledgements
We are greatly indebted to the patients included in the study. We thank V. Cotard, C. Goutsmedt, M.-F. Le Cunff and N. Bourgeais for assistance in patient recruitment and L. Beekman for his technical support. We thank the biological resource centre for biobanking (CHU Nantes, Nantes Université, Centre de ressources biologiques (BB-0033-00040), F-44000 Nantes, France) for applying the following guidelines68. We are most grateful to the Genomics and Bioinformatics Core Facility of Nantes (GenoBiRD, Biogenouest, IFB) for its technical support. This research has been conducted using the UK Biobank resource; we are grateful to UK Biobank participants. The MINE study (J.H.V.) has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 772376—EScORIAL). The collaboration project is cofunded by the PPP Allowance made available by Health~Holland, Top Sector Life Sciences & Health, to stimulate public–private partnerships. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113, 085475 and 090355. The KORA research platform (KORA, Cooperative Research in the Region of Augsburg) was initiated and financed by the Helmholtz Zentrum München—German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. J. Barc is supported by the research program Etoiles montantes des Pays de la Loire REGIOCARD RPH081-U1087-REG-PDL, ANR JCJC LEARN (R21006NN, RPV21014NNA) and by the H2020-MSCA-IF-2014 Program of the European Commission (RISTRAD-661617). R.T. is supported by the Canadian Heart Rhythm Society’s George Mines Award, the European Society of Cardiology research award, and the Philippa and Marvin Carsley Cardiology Chair. D.Y.C. is supported by Fondation Leducq and National Institutes of Health (NIH) NHGRI T32 (no. 1T32HG010464-01). M. Baudic was supported by IRP—VERACITIES—New Mechanisms for VEntricular ARrhythmia And CardIomeTabolic DIseasES, an I-SITE NExT health and engineering initiative (Ecole Centrale and Nantes University) and by the IRP—GAINES—Genetic Architecture IN cardiovascular disEaSes funded by INSERM and CNRS. R.W. is supported by an Amsterdam Cardiovascular Sciences fellowship. S.C. is supported by the NHLBI BioData Catalyst Fellows Program. C.A.R. is supported by Fondation Leducq, the Dutch Heart Foundation (CVON PREDICT2) and the Innovational Research Incentives Scheme Vidi grant from the Netherlands Organisation for Health Research and Development (ZonMw; 91714371). Y.D.W. is supported by the Robert Lancaster Memorial Fund. M.P. is supported by Cardiac Risk in the Young. S.V.D. is supported by Wetenschappelijk Fonds Willy Gepts VUB-UZ Brussel, project ‘Unravelling the molecular genetic pathways of Brugada Syndrome by cardiomics research’, VUB IRP project ‘IMAGica: an Integrative personalized Medical Approach for Genetic diseases, Inherited Cardia Arrhythmias as a model’ and Innoviris BRIDGE 2017, project ‘IGenCare: Integrated Personalised Medical Genomics Care Solution for Patients with Rare Genetic Diseases’. S.H. is supported by the Barts BRC. B.R. is supported by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research). B.G.W. is supported by the Danish Heart Foundation. M.B.S. is supported by K23HL127704. Project MinE Belgium was supported by a grant from IWT (no. 140935), the ALS Liga België, the National Lottery of Belgium and the KU Leuven Opening the Future Fund. D.C. and C.L. are supported by HYPERGENES (HEALTH-F4-2007). D.R. is supported by R01 HL149826, P50 GM115305. P.J.S. acknowledges the support of Leducq Foundation for Cardiovascular Research grant 18CVD05. P.V.D. is supported by the Netherlands CardioVascular Research Initiative (CVON PREDICT2). C.A. is supported by NIH HL47678 and HL138103, W.W. Smith Charitable Trust and Wistar Morris Fund. M.B. is Supported by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research). P.D.L. is supported by UCL/UCLH Biomedicine NIHR and Barts BRC. B.L. is supported by GOA—Antigone 33933. J.B. is supported by a Senior Clinical Fellowship of the Flemish Science Foundation (FWO). E.B. is supported by the British Heart Foundation including BHF Clinical Research Training Fellowship (FS/11/71/28918: Future diagnostic role and new genetic loci in SADS), Cardiac Risk in the Young and Robert Lancaster Memorial fund sponsored by McColl’s Ltd. Retail Group. H.L.T. is supported by the European Union’s Horizon 2020 research and innovation program under acronym ESCAPE-NET, registered under grant agreement no. 733381, and the Dutch Heart Foundation (CVON RESCUED and PREDICT2 projects). M.D. is supported by NIH-RO1 HL134328. P.T.E. was supported by the Fondation Leducq (14CVD01), the NIH (1RO1HL092577, R01HL128914, K24HL105780), the American Heart Association (18SFRN34110082) and by a research grant from Bayer AG to the Broad Institute. S.A.L. is supported by NIH grant 1R01HL139731 and American Heart Association 18SFRN34250007. J.-B.G. received a grant from the Fédération Française de Cardiologie (PREVENT project). A.L.G. is supported by the Fondation Leducq. C.A.M.R. is supported by the Leducq Foundation and Burroughs Wellecome Fund. A.A.W. is supported by the Dutch Heart Foundation (CVON PREDICT2 project). J.-J.S. is supported by the Fondation pour la Recherche Médicale (DEQ20140329545). R.R. and P.G. are supported by the National Agency for Research (ANR-GENSUD-14-CE10-0001). C.R.B. is supported by the Dutch Heart Foundation (CVON PREDICT2 project), the Netherlands Organization for Scientific Research (VICI fellowship, 016.150.610) and Fondation Leducq (17CVD02).
Author information
Authors and Affiliations
Consortia
Contributions
J. Barc, R.T., C. Glinge, D.Y.C., M.J., J.-J.S., P.W.B., A.L.G., C.A.M.R., C.D., M.W.T., R.R. and C.R.B. conceived/designed elements of the study. All authors acquired, analyzed or interpreted data. J. Barc, R.T., C. Glinge, D.Y.C., M.J., A.O.V., J.-J.S., P.W.B., A.L.G., C.A.M.R., C.D., M.W.T., A.A.W., R.R. and C.R.B. drafted the manuscript. All authors critically revised the manuscript for important intellectual content and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
P.V.D. holds a senior clinical investigatorship of FWO-Vlaanderen and is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders. S.A.L. receives sponsored research support from Bristol Myers Squibb/Pfizer, Bayer AG, Boehringer Ingelheim and Fitbit, and has consulted for Bristol Myers Squibb/Pfizer and Bayer AG, and participates in a research collaboration with IBM. P.T.E. has served on advisory boards or consulted for Bayer AG, Quest Diagnostics, MyoKardia and Novartis. A.L.G. is part of the Scientific Advisory Board for Amgen, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Marcel den Hoed and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Developmental transcription factor gene enrichment.
In 10,000 simulations of 12 SNP sets matching the non-chromosome 3 BrS lead SNPs (permutation-based unilateral (one-sided P-value) enrichment test), no more than six DNA binding protein genes were located within 300 kb (range: 0-6 genes). In contrast, among the 12 non-chromosome 3 lead SNPs, 10 overlapped or were located within 300 kb of DNA binding protein genes (P = 1 × 10−4). DNA binding protein genes were defined according to number of Gene Ontology: 0043565.
Extended Data Fig. 2 Annotation of the chromosome 18 locus.
Top panel, Locus Zoom plot of BrS associated variant at the chromosome 18 locus based on the meta-analysis data. –log10 P-values are shown along the left y-axis, and the right y-axis corresponds to recombination rate, plotted as a blue line. The x-axis indicates chromosomal position. The lead variant at each association signal is shown as a purple diamond. Other variants are colored according to their correlation (r2) with the lead variant (see legend). Middle panel, Graphic representation of the chromosome 18 locus displaying the following annotations: genomic coordinates (hg19), topologically associated domains (TAD)69; region containing SNPs in linkage disequilibrium with the lead SNP at the locus (red bar, LD, r² ≥ 0.6); genes located in the locus; chromatin interactions identified in human left ventricular tissue51 overlapping the association signal (± 40 kb corresponding to the Hi-C dataset resolution), and expression quantitative trait loci (eQTL) reported in GTEx20 for human left ventricular tissue. Bottom left panel, eQTL violin plot from GTEx displaying significant association between the lead SNP of the chromosome 18 locus and the expression of MAPRE2 within the cardiac ventricular tissue. The number of left ventricular heart samples per genotype is shown in parentheses. In the box plots, the middle line shows the median value of the expression of each genotype, while the violin plots show the distribution. Bottom right panel, Locus compare plot depicting SNPs at the chromosome 18 locus. The eQTL –log10 P-value is plotted on the y-axis, and the BrS GWAS meta-analysis –log10 P-value is plotted on the x-axis. SNPs are colored according to their degree of linkage disequilibrium (r2), with the lead BrS associated variant highlighted as a purple diamond. eQTL, expression quantitative trait locus. Note that a different scale is used in the top and middle panels.
Extended Data Fig. 3 Cellular electrophysiology studies in MAPRE2 knockout and control hiPSC-CMs.
Typical current recordings and (in)activation properties in control (CTRL) human induced pluripotent stem cell derived cardiomycytes (hiPSC-CMs) and without MAPRE2 (MAPRE2 KO). a, Typical sodium current (INa) in a CTRL and MAPRE2-KO hiPSC-CM. b, Voltage-dependency of activation (squares) and inactivation (circles) in CTRL and MAPRE2 KO hiPSC-CMs. The V1/2 of voltage dependency of activation was 4.2 mV more positive (P = 0.0016) in MAPRE2 KO compared with CTRL hiPSC-CMs. c, Recovery from inactivation in CTRL and MAPRE2 KO hiPSC-CMs. d, Typical repolarizing outward currents (IOutward) in a CTRL and a MAPRE2 KO hiPSC-CM. e, Voltage-dependency of IOutward activation in CTRL and MAPRE2 KO hiPSC-CMs. CTRL, control; KO, knock-out. Data in b, c, and e are presented as mean values ± s.e.m. Statistical test used was two-sided unpaired t-test.
Extended Data Fig. 4 Distribution of PRSBrS in specific patient sub-groups.
Histograms displaying distribution of PRSBrS in specific patient sub-groups. a, PRSBrS distribution comparing SCN5A– BrS cases presenting with a spontaneous type 1 BrS ECG with SCN5A– BrS cases presenting with a type 1 BrS ECG after sodium channel blocker challenge (drug-induced). b, PRSBrS distribution comparing SCN5A+ BrS cases presenting with a type 1 ECG at baseline with SCN5A+ BrS cases presenting with a type 1 BrS ECG after sodium channel blocker challenge. PRSBrS was calculated per individual based on the 21 BrS risk alleles and their corresponding effect sizes. Results were obtained after logistic regression, two-sided P-value not corrected for multiple testing. Reported P-values refer to the difference in PRSBrS units between two groups.
Extended Data Fig. 5 Comparison of effect on BrS susceptibility with effect on ECG intervals (PR, QRS and QT) and AF susceptibility from previously published GWAS.
a-d, Effect size of the BrS risk alleles compared to their respective effect size on PR-interval (a), QRS-duration (b), QT-interval (c) and AF risk (d). In all panels, the x-axis represents the effect on BrS susceptibility from the BrS GWAS meta-analysis, presented either as log(odds ratios) with 95% confidence intervals (a-c), or as odds ratios with 95% confidence intervals (d). For a-c, the y-axis represents the effect estimates and 95% confidence interval (milliseconds per allele) calculated for PR-interval, QRS-duration or QT-interval in previously published GWAS conducted in the general population4,5,6. For d, the y-axis represents the effect on AF susceptibility from previously published GWAS7, shown as odds ratios with 95% confidence intervals. Red color indicates that the BrS risk allele (or a proxy with r2 > 0.8) is associated with a prolongation of the ECG parameter or an increased risk of AF in the published GWAS at P < 1 × 10−5, while blue color indicates that the BrS risk allele is associated with a shorter ECG interval or a decreased risk of AF at P < 1 × 10−5. Loci annotated with a gene name in grey reached at least nominal significance in the published GWAS (P < 0.05). BrS, Brugada syndrome; AF, atrial fibrillation or flutter; PR, PR-interval; QRS, QRS-duration; QT, QT-interval; OR, odds ratio. Effect sizes of all 21 BrS susceptibility SNPs or their proxies on PR-interval, QRS-duration, QT-interval and AF risk in previous GWAS studies are listed in Supplementary Tables 14–17.
Supplementary information
Supplementary Information
Supplementary Note, Figs. 1–9 and Tables 1–19.
Supplementary Table 1
Supplementary Tables 1–19.
Rights and permissions
About this article

Cite this article
Barc, J., Tadros, R., Glinge, C. et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat Genet 54, 232–239 (2022). https://doi.org/10.1038/s41588-021-01007-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-01007-6
This article is cited by
-
Targeted phasing of 2–200 kilobase DNA fragments with a short-read sequencer and a single-tube linked-read library method
Scientific Reports (2024)
-
Stem cell models of inherited arrhythmias
Nature Cardiovascular Research (2024)
-
The leading example of the Leducq Foundation
Nature Cardiovascular Research (2024)
-
Postmortale Genetik nach einem plötzlichen Herztod
Herzschrittmachertherapie + Elektrophysiologie (2024)
-
Kardiogenetik in Deutschland – ein (Rück‑)Blick
Herzschrittmachertherapie + Elektrophysiologie (2024)
