
Abstract
The coronavirus disease 2019 (COVID-19) pandemic has affected the world radically since 2020. Spain was one of the European countries with the highest incidence during the first wave. As a part of a consortium to monitor and study the evolution of the epidemic, we sequenced 2,170 samples, diagnosed mostly before lockdown measures. Here, we identified at least 500 introductions from multiple international sources and documented the early rise of two dominant Spanish epidemic clades (SECs), probably amplified by superspreading events. Both SECs were related closely to the initial Asian variants of SARS-CoV-2 and spread widely across Spain. We inferred a substantial reduction in the effective reproductive number of both SECs due to public-health interventions (Re < 1), also reflected in the replacement of SECs by a new variant over the summer of 2020. In summary, we reveal a notable difference in the initial genetic makeup of SARS-CoV-2 in Spain compared with other European countries and show evidence to support the effectiveness of lockdown measures in controlling virus spread, even for the most successful genetic variants.
Similar content being viewed by others


Main
The new coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) emerged in China in October–November 2019 (ref. 1) and by the end of March 2020 it was present in most countries of the world. The World Health Organization (WHO) declared the new disease as a pandemic on 11 March 2020. Spain suffered a severe epidemic, with the first case reported on 29 January 2020 (ref. 2), and an accumulated number of 249,659 cases by 1 July 2020, including 28,363 fatalities3. Furthermore, a nationwide seroprevalence study showed that only one in ten cases of infection by SARS-CoV-2 were diagnosed and reported in that period4, suggesting that the total number of infections has been vastly underestimated. Spain ordered a series of nonpharmaceutical intervention measures, including a general lockdown on 14 March 2020 (ref. 5), later applied by many other countries, and was successful in reducing infection rates by the end of May 2020 (ref. 6). Despite these measures, almost 30,000 individuals died during the first wave of the epidemic (until 14 May 2020), and a second wave of COVID-19 was beginning to emerge by the beginning of July 2020 (ref. 7).
Despite the high incidence of infection across the country, some regions had substantially higher incidence than others. Genomic epidemiology and phylodynamics8,9,10 offer a unique opportunity to understand the early events of the epidemic at the global, regional and local levels, to track the evolution of the epidemic after its initial stages and to quantify the impact of lockdown measures on the genetic variants of the virus. However, there are challenges and caveats that prevent the use of pathogen genomes as the sole source of interpretation. While there is now a large number of SARS-CoV-2 sequences deposited in GISAID11, there are still important unsampled areas of the world, including some that played an important role in the initial spread of the epidemic. In addition, the virus spreads faster than it evolves12,13 which limits the resolution of phylogenetic and phylodynamic analyses14. Finally, despite important efforts by sequencing consortiums, only a fraction of the total number of infections has been sequenced. Nevertheless, genomic epidemiology has played an important role in understanding the global and local epidemiology of COVID-19 (refs. 15,16,17).
After the pandemic was declared in Spain, we assembled the National Consortium of genomic epidemiology of SARS-CoV-2 (http://seqcovid.csic.es/). This established a unique network incorporating more than 50 hospitals and scientific institutions across the country to collect clinical samples and epidemiological information from COVID-19 cases. Here, we present the results of this nationwide effort. We were able to sequence 12% of the reported cases before the national lockdown, and 1% of the reported cases of the first wave when lockdown measures ended (14 May 2020), including samples of SARS-CoV-2 across Spain in the early months of the pandemic (February–May 2020). Using a combination of pathogen genomics, phylogenetic tools and clinical and epidemiological data, we have been able to dissect the very early events in the dispersion of SARS-CoV-2 throughout Spain, as well the evolution of the virus during the exponential phase and after the lockdown. We document simultaneous introductions into the country from multiple sources. We show that up to 40% of cases were caused by two SECs, named SEC7 and SEC8. Seven other SECs were detected but their role was minor, probably because they were introduced relatively close to the lockdown and, unlike the initial two clades, had no opportunity for a rapid exponential expansion. In contrast to clades from other European countries, these SECs belong to early lineages in the epidemic (A in Pango, 19B in NextStrain). We also show that the reproductive number, Re, of the most successful SECs declined quickly after the implementation of lockdown measures, and they were completely absent from samples taken in July–September 2020. Our results suggest that the most successful variants were those associated with earlier introductions, but also that their success may have depended on the synergy between superspreading events and high mobility. These results also show the effectiveness of lockdown measures in controlling the virus spread and eliminating established successful epidemic clusters from circulation.
SARS-CoV-2 was introduced multiple times from multiple sources
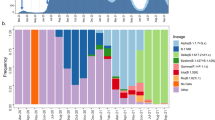
Our dataset consists of 2,170 sequences from Spain, collected under ethical approval, from 25 February to 22 June 2020, coinciding with the initial phases of the COVID-19 pandemic in the country (Fig. 1a). The most populated Spanish regions were sampled, resulting in a dataset with sequences representing 16 of the 17 administrative regions into which the country is divided (Fig. 1b). Of the 2,170 (90.4%) samples analyzed here, 1,962 have been sequenced by the SeqCOVID consortium, while the remaining 208 have been generated by independent laboratories and downloaded from GISAID11 (Supplementary Table 1). Spain showed a particular viral population structure with a higher proportion of lineage A sequences compared with other European countries18 (Fig. 1c). Strains from patients in Spain were related more closely to strains from cases sequenced in China, and were the most abundant during the first weeks of the epidemic in Spain. They were later replaced by lineage B strains (Extended Data Fig. 1), which differ by at least six to seven substitutions from lineage A and that dominated the beginning of the pandemic in most European countries, in contrast to the patterns seen in Spain. In addition, we observed a heterogeneous distribution of the SARS-CoV-2 genetic diversity within Spain, both at regional and local levels. For example, our analysis shows that viral diversity was higher in some urban areas, and it declined with geographic distance from the city centers, as observed in Valencia (Supplementary Note).

a, Distribution of sequenced samples (blue) versus confirmed cases in Spain (gray) by date. Country lockdown measures were in effect from 14 March to 14 May 2020. b, Distribution of the sequenced samples across Spain plotted in Microreact. These data can be explored with more detail in the Microreact webpage (https://microreact.org/showcase) loading the Supplementary Data 1 files. The size of each piechart correlates with the number of sequences collected in the corresponding area. Each color corresponds to a specific Pango lineage, as detailed in Extended Data Fig. 1 (light yellow and green correspond to lineage A, all the others are lineage B). The map image was extracted from the Microreact visualization37. c, Distribution of main SARS-CoV-2 clades during the first stages of the pandemic (before 1 April 2020), in those European countries with more than 50 sequences deposited in GISAID as of 13 November 2020. d, Global ML phylogeny constructed with 32,416 sequences, placement of Spanish samples is indicated in red. e, New and accumulated introductions to Spain. Lower-bound introduction estimates were defined as the probable date of the infection of the first case in a cluster (14 days before symptom onset). f, Estimated international origin of SARS-CoV-2 introductions based on phylogenetic data; in color, those countries with a probable contribution greater than 10%.
Phylogenomic analyses suggest the existence of multiple independent entries of the virus into Spain, similar to what was seen for other countries19,20. To identify possible introductions, we inspected the placement of Spanish viral samples in a global phylogeny constructed with more than 30,000 sequences (Fig. 1d). Given the low genetic diversity of the virus, particularly at the beginning of the epidemic, we found many instances in which a Spanish sample was genetically identical to other variants circulating in the rest of the world. According to their phylogenetic placement, three different possibilities were considered for the phylogenetic position of Spanish sequences. A sequence was included in a ‘candidate transmission cluster’ when it was found in a monophyletic clade with other Spanish sequences; it was included in a ‘zero-distance’ group when it grouped with other genetically identical Spanish sequences but also with other foreign sequences; and it was denoted as ‘unique’ when no matching sequence in the Spanish dataset was identified and the sequence differed by more than one single nucleotide polymorphism (SNP) from other Spanish sequences (Supplementary Fig.1a; detailed definitions of the groups are in Methods and Extended Data Fig. 2). We detected 224 ‘candidate transmission clusters’, comprising 827 sequences (~40% of the Spanish samples); 30 ‘zero-distance clusters’, comprising 831 sequences, and 513 ‘unique’ sequences (Supplementary Fig. 2). Next, we determined how many unique cases and clusters were compatible with an introduction before the general lockdown. We detected that 191 groups (165 ‘candidate transmission clusters’ plus 26 ‘zero-distance clusters’) and 328 unique sequences met this criterion, representing at least 519 independent introductions (distribution of dates in Fig. 1e, distribution of ‘unique’ sequences across regions in Supplementary Fig. 1b). This is probably an underestimate of the total number of entries because the number of sequences analyzed is a small subset of the total notified cases (Fig. 1a). Phylogenetic analysis suggests that the most probable introduction of cases with a clear phylogenetic link (Methods) came from Italy, the Netherlands, England and Austria (accounting for ~23%, ~20%, ~13% and 12% of the cases for which a probable country of origin can be inferred, respectively) (Fig. 1f). The observation that more than half of the introduction events detected are unique sequences illustrates the disparate outcomes after an introduction, as some events resulted in large epidemiological clusters, and others disappeared leaving almost no trace. A clear example is the case of the first described death in Spain, for which we have generated a partial viral sequence. The patient was infected in Nepal but there were no identifiable secondary cases in our dataset.
A few genetic variants dominated the first wave in Spain
To identify those introductions that resulted in sustained transmission and, therefore, the ones that were epidemiologically successful in the long term, we scanned the phylogeny for larger clades comprised mainly of Spanish samples (Methods). We identified nine SECs distributed across the phylogeny, representing 46% of the total Spanish dataset analyzed (995 out of 2,170 samples) (Fig. 2a, Extended Data Figs. 3 and 4 and Supplementary Tables 1 and 2). We noticed first that only two SECs encompassed 30% and 10% of all Spanish samples (SEC8 and SEC7, respectively). This implies that the introduction of these two specific genetic variants explains a high proportion of the entire epidemic for the first wave in the country. In fact, they were responsible for 44% of the ‘candidate transmission clusters’ identified before the lockdown (Fig. 2b). We then estimated the time of introduction in Spain for the nine SECs using a Bayesian approach (Supplementary Table 2). As a conservative estimate we considered the time of introduction as any time between the age of the most recent common ancestor of the SEC and the date of the first Spanish sample (Fig. 2c). Thus, we assume that the ancestor of the SEC was not necessarily in Spain.

a, ML phylogenetic tree of Spanish sequences indicating the identified SECs (inner circle), the Pango lineage (middle circle) and the NextStrain clade (outer circle). b, Range of dates for each ‘candidate transmission cluster’ identified within the SECs, and the most probable origin date (14 days before the first documented case). c, Time of the MRCA of each SEC is plotted, including the 95% HPD interval. First collected sample, and whether it is Spanish or non-Spanish, is indicated. d, SEC dispersion through the different regions of the country. Some SECs are restricted to one or two regions, while others have expanded through the complete territory.
Our analysis shows that the earlier the introduction, the larger the size of the SEC (Supplementary Fig. 3). The larger clades, SEC7 and SEC8, were the first successful genetic variants introduced into Spain during late January–February 2020 (Fig. 2b). Both belong to lineage A (Pango nomenclature) and partially explain the particular population structure observed in Spain relative to other European countries (Fig. 1c). In addition, compared with other SECs, SEC7 and SEC8 were spread widely in the country, being present in at least 10 of the 17 administrative regions (Fig. 2b), and having a mean pairwise geographic distance between samples of more than 300 km, regardless of whether or not the Islas Canarias and Baleares are included (Extended Data Fig. 5). By contrast, SECs that were introduced later were smaller and showed a narrower geographic spread (between 0 and 58 km, analysis of variance (ANOVA)-adjusted P value « 0.01, Supplementary Note).
Superspreading events and mobility led to success of SEC8
Why some genetic variants succeed over others cannot be answered from genomic sequence data alone. We must also consider the epidemiological dynamics in the country. There are data supporting a role for the spike protein mutation 614G in epidemiological success. However, SEC7 and SEC8 do not harbor that mutation, explaining why 614G was less frequent in Spain during the first weeks of the epidemic than in other countries (Extended Data Fig. 6). In addition, analysis of signature positions for both SECs did not lead to any probable genomic determinant of epidemiological success (Supplementary Table 3). Unfortunately, we had no access to linked epidemiological data for the complete dataset. However, we had access to detailed information from two superspreading events linked to SEC8. On the basis of the phylogenetic analysis and the linked epidemiological data, we are able to shed light on the early success of SEC8, the dynamics of which can be explained in three stages.
In the first stage, SEC8 was introduced at least twice from Italy to the city of Valencia (Fig. 3a). There is epidemiological evidence that the individual in both cases became infected in Italy, as they attended the Atalanta-Valencia Champions League football match on 19 February 2020, and that, upon returning to Valencia a few days later, one of them initiated a transmission chain of at least 24 cases according to Public Health sources. Of these 24 cases, 12 were sequenced (Fig. 3a, highlighted in orange). This epidemiological link strongly suggests that the SEC8 genetic variant was imported from Italy. This introduction occurred in agreement with the estimated time of entry of SEC8 into Spain (Supplementary Table 2). NextStrain tracking tools for viral spatial spread suggest additional SEC8-related early seedings in Madrid, País Vasco, Andalucía and La Rioja regions (Supplementary Data 2), which might have involved other countries, not exclusively Italy. Given the lack of genetic differentiation of the virus and scarce epidemiological information, there is no certainty on whether these infections resulted from independent introductions from abroad or from internal migrations of infected persons, although the simultaneous detection in different regions favors the first option. Most of these multiple introductions occurred during the second half of February 2020, a period in which more than 11,000 daily entries of travelers from Italy were recorded.

a, ML phylogeny with all strains of SEC8. Samples with epidemiological evidence about their origin are marked in the tree. In red, cases imported from different events in Italy. In orange, secondary cases originated from one of the cases introduced from Italy (also marked with blue arrows). In purple, cases related to a large funeral in La Rioja. Green stars mark potential superspreading events of more than ten sequences sharing at least one clade-defining SNP. b, Contribution of SEC8 to the total of samples sequenced over time. The horizontal red line marks the start of the Spanish lockdown on 14 March 2020. c, Phylodynamic estimates of the reproductive number (Re) of SEC8. The x axis represents time, from the origin of the sampled diversity of SEC8 to the date of the last collected genome on 16 May 2020. The blue dotted line shows the posterior value of the timing of the most important change in Re, around 9 March 2020 [95% HPD: 8–10 March]. The y axis represents Re, and the violin plots show the posterior distribution of this parameter before and after the change time in Re, with a mean of 3.14 [95% HPD: 2.71–3.57] and 0.23 [95% HPD: 0.15–0.32] before and after the change time, respectively. The phylogenetic tree in the background is a maximum clade credibility tree with the tips colored according to whether they were sampled before or after 9 March 2020. The lower whisker, higher whisker, center and bounds of each boxplot refers to quartile 1–1.5 interquartile range, quartile 3 + 1.5 interquartile range, mean, first and third quartiles of the data. Individual points are outliers (values lower than quartile 1–1.5 interquartile range and higher than quartile 3 + 1.5 interquartile range). Boxplot was constructed with all the Spanish sequences in SEC8 (N = 636).
In a second stage, SEC8 was fueled by superspreading events (Supplementary Data 2). On the basis of the topology of the phylogenetic tree (Fig. 2a), there were multiple clades within SEC8 involving a large number of very closely related sequences (1–3 SNPs) (Fig. 3a). Of special relevance was a funeral on 23 February 2020, with attendees from the País Vasco and La Rioja regions (Public Health officers estimated 800 attendees, resulting in 36 confirmed symptomatic cases) from which 25 samples were sequenced successfully (Fig. 3a, highlighted in purple). Importantly, although they did not differ by more than two SNPs, these sequences are spread across the SEC8 phylogeny, suggesting the existence of many more nonsampled secondary cases across the country (Fig. 3a). In a third stage, SEC8, after reaching high frequencies locally, was redistributed across the country and, in less than 2 weeks, it reached a prevalence of 60% among the sequenced genomes (Fig. 3b), being present in almost every region analyzed. All these stages occurred between the first known diagnosed SEC8 case on 25 February 2020 (Supplementary Table 2) and before the lockdown on 14 March 2020, highlighting the need for very early containment measures to stop the spread of SARS-CoV-2.
Effect of lockdown on the main clades
In the second half of March 2020, Spain imposed a strict lockdown on nonessential services and movements. Consistently, the number of cases for all SECs dropped quickly after the lockdown (Extended Data Fig. 7). A Bayesian birth–death skyline analysis allowed us to evaluate the effect of the lockdown on the effective reproductive number (Re) of the most successful SECs. The analyses of SEC7 (Extended Data Fig. 8) and SEC8 (Fig. 3c) resulted in similar estimates for Re before the lockdown (2.10 with 95% highest posterior density (HPD):1.67–2.62 and 3.14 HPD: 2.71–3.57, respectively) similar to the Re estimated early in the epidemic for SARS-CoV-2 (ref. 21,22). After the lockdown there was a substantial decrease to less than 0.5 in both cases (0.27 95% HPD: 0.06–0.47; 0.23 HPD: 0.15–0.32, respectively). The model also estimated that the date with highest support for a change in Re coincides roughly with the start of the lockdown in Spain on 14 March 2020 (20 March HPD: 15–25 March; 9 March HPD: 8–10 March, respectively). In addition, we calculated the doubling time for both SECs23. Before the corresponding date of change for Re, the doubling time for SEC7 was estimated at 6.3 days (95% HPD: 4.3–10.2 days) and that for SEC8 at 3.3 days (95% HPD: 2.7–4.1 days). Re values after those dates had a posterior distribution that did not include 1.0 for both SECs (Supplementary Note), a result that supports the reduction in the rate of increase of confirmed cases and that is in agreement with estimates from epidemiological models and data21,22.
In addition, all the viral variants not included in the SECs, mostly harboring the 614G mutation, displayed a similar decrease in the number of cases after the lockdown compared with the most successful SECs (Extended Data Fig. 9a). The impact of the measures implemented in the Re was also evaluated in two representative Pango lineages carrying the 614G mutation, and a substantial decrease in Re after the lockdown was observed, from an Re greater than 1.5 to an Re of ~0.25, similar to the results obtained for SEC7 and SEC8 (Extended Data Fig. 9b).
Discussion
Our analyses have revealed more than 500 independent introductions of SARS-CoV-2 into Spain between late January, coinciding with the first reported cases in our country2,24, and mid-April 2020. The earliest entries corresponded to lineage A, matching the virus diversity profile reported for the country. This lineage was common in Asia but rare in the rest of Europe25. We observed that two genetic variants (SEC7 and SEC8) of this lineage dominated the first stages of the epidemic wave in Spain, contrary to what was observed in other European countries. In fact, most cases described in Europe at the beginning of the pandemic were lineage B, which makes the situation in Spain more unique. This highlights the importance of epidemiological data, from which we know that SEC8 was introduced at least from Italy despite not being the dominant lineage in the country at that time and illustrating the role of founder effects early in the pandemic18,26,27.
Reasons why some variants dominate over others can be related to viral genetics, to founder events associated to particular variants and to the implementation of different public health measures over time, not necessarily in an exclusive manner. The variant distribution could also be explained partly by sampling bias. No mutation probably associated with epidemiological success has been identified in our analyses of SEC7 and SEC8 (Supplementary Table 3). In fact, neither SECs carry the 614G mutation in the spike protein, contrary to what is seen in most lineage B variants (Extended Data Fig. 6). The mutation 614G has been associated with increased viral shedding compared to the ancestral 614D variant in laboratory conditions28 and in transmission studies29,30. Consistently, our analysis supports the observation30 that 614G strains had higher associated viral loads measured as lower cycle threshold (Ct) values (Supplementary Fig. 4). However, one study reports that its actual role in the epidemic is doubtul31, suggesting that its impact, if any, on epidemic transmission was minor. In the case of Spain, 614G did not account for the initial success of the epidemic because SEC7 and, in particular, SEC8 were much more common than other genetic variants until the lockdown (10% and 30% of cases, respectively). Notably, 614G strains were introduced and expanded later, and closer to the start of lockdown, than 614D (Extended Data Figs. 6 and 9a), explaining the particular lineage structure observed in Spain (Fig. 1c). By contrast, founder events seem to have played an important role for the two main SECs. Our analysis shows that these two SECs were the first variants introduced in the country and, at least in the case of SEC8, were linked to very early superspreading events that contributed to their success. However, an early introduction of lineage A variants also occurred in other European countries, but they did not take hold and were displaced sooner by lineage B. Despite the early adoption of strong nonpharmaceutical intervention measures, we hypothesize that epidemic control in the first wave in Spain was soon overwhelmed as compared with countries that controlled early outbreaks15. This was probably associated with a strict implementation of the case definition by the WHO, which allowed for a stealth dispersion of the first introductions. Spain implemented one of the strictest lockdowns in Europe, with a high compliance from the population as tracked by mobility data32. The efficacy of nonpharmaceutical intervention measures was evident a few weeks later, and it was reflected in the almost complete elimination of SEC7, SEC8, and most variants by the end of the first wave (Extended Data Figs. 7 and 9a). However, we do observe a replacement of lineage A (SEC7 and SEC8) just after the start of the lockdown by B.1 variants harboring D614G (Extended Data Fig. 9a). Contrary to that of lineage A, the spread of B.1 variants in Spain was represented by smaller SECs and more isolated cases. It is probable that these smaller clusters correspond to a new stage in the epidemic at the national level characterized by more limited mobility and social interactions.
This study has several limitations. Even though Spain is one of the countries with high contribution to public repositories, our dataset represents only a small subset of confirmed cases that occurred in the first COVID-19 wave (1% of cases). Moreover, sampling across the country was heterogeneous and the representation of each region in the dataset was not always proportional to the incidence during the studied period. Lack of genome data from countries with high disease burden, especially at the beginning of the pandemic, may have prevented a reliable identification of their probable sources based only on viral genome sequences. In addition, we did not have access to individual patient data for most cases. These caveats could have an impact on, for example, an exact quantification on the number of introductions, which will be always an estimate. However, our analysis already gives clues about the role of multiple introductions in the early days of the epidemic. Despite these limitations, we have been able to investigate some of the key cases and events that initiated the epidemic in Spain. This allowed us to understand the origin and early spread of SEC8, which would not have been possible based only on genome data. But we have also shown that genetic data can be used to accurately estimate relevant epidemiological parameters, such as Re and doubling times, even when the proportion of sampling is low.
We believe that our results allow us to draw lessons for the control of this, as well as future, pandemics. First, we have shown how specific variants can be used to track the effectiveness of public health control measures. In February 2020, the number of SEC8 cases was just a few dozen and yet it ended up accounting for 60% of the sequenced samples in the first weeks of March 2020. Second, the closure of borders to countries with high incidence is relevant to reduce simultaneous and multiple imports of the virus, but the efficacy of these restrictions also depends on the internal incidence of the disease33. The most successful SECs during the first wave were probably those that arrived early, several times, and to diverse locations. Thus, as suggested elsewhere36, founder effects are important for the success of certain variants. Third, SEC7 and SEC8 spread across Spain in a matter of days. Controlling mobility is essential when the level of community transmission is high, as demonstrated by the important decrease in Re for these high-transmission genetic variants after the lockdown. As a comparison, before the lockdown, Re values were 50% higher in Spain (3.3 for SEC8) than in Australia (1.63), and they underwent a reduction down to 7% of the original value (0.23) as a result of the containment measures, compared to a reduction to 30% (0.48) in Australia17. From a public health perspective, our results add to the evidence that the success of specific genetic variants, with no intrinsic biological difference, is fueled by superspreading events that rapidly increase the prevalence of the virus34. Subsequently, coupled to the high mobility of our connected world, a variant may end up dominating the epidemic in a particular geographic location. This is what occurred with SEC8 and what, at a local level, has been described in Boston35. In fact, we have recently described a new variant in Europe, the prevalence of which was growing rapidly in several countries during the summer of 2020, which is also linked to initial superspreading events36. By contrast, new variants with different transmissibility and immunogenicity profiles started to merge at the end of 2020. Some of these variants, such as B.1.1.7 (alpha) were able to replace existing variants in a matter of months. For the first wave in Spain, the conclusion is that early diagnosis and notification of cases would have helped timely implementation of effective contact tracing that, coupled with earlier mobility closures and maybe tighter border control, could probably have delayed by a few days the expansion of genetic variants, such SEC8, during the early stages of the epidemic. Whether this might have changed the global shape of the epidemic in the country or whether other genetic variants would have performed this role instead, leading to a similar outcome, cannot be ascertained. The comparison with other countries leads us to suspect that the difference would have been minimal.
Methods
SeqCOVID sampling and sequencing
RNA samples were received from different hospitals, and confirmed as SARS-Cov-2-positive by PCR with reverse transcription (RT-PCR) by Microbiological Services. Samples consisted of the remaining RNA extracts from naso- and oropharyngeal clinical specimens used for diagnosis. The use of such samples has been approved by the ethics committee Comité Ético de Investigación de Salud Pública y Centro Superior de Investigación en Salud Pública (CEI DGSP-CSISP) N° 20200414/05.
In general, we applied the following criteria for selecting the samples that underwent sequencing: (1) only one sample per patient, (2) diagnostic PCR should have a Ct under 30, (3) prioritize samples from poorly sampled regions and hospitals to maximize geographic diversity, (4) prioritize samples according to their diagnosis date, to maximize sampling of high incidence periods. These criteria were adopted weeks before the beginning of the project, after analyzing the firsts sets of sequences, so, in the initial weeks, we did not preselect the samples for sequencing.
In the SeqCOVID consortium webpage (http://seqcovid.csic.es/), the number of samples received, sequenced and uploaded to public repositories are shown and updated periodically.
RNA was retrotranscribed into cDNA and SARS-CoV-2 complete genome amplification was conducted in two multiplex PCRs, accordingly to an openly available protocol developed by the ARTIC network38 using the V3 multiplex primers scheme39. Two resulting amplicon pools were combined and used for library preparation. Genomic libraries were constructed with the Nextera DNA Flex Sample Preparation kit (Illumina Inc.) according to the manufacturer’s protocol, with five cycles for indexing PCR. Whole genome sequencing was carried out in the MiSeq platform (2 × 200 cycles paired-end run; Illumina).
The sequences obtained went through a bioinformatic pipeline based on IVAR40, which is open source and can be accessed at https://gitlab.com/fisabio-ngs/sars-cov2-mapping. In short, the pipeline goes through the following steps: (1) Removal of the human reads with Kraken41; (2) filtering of the fastq files using fastp v.0.20.1 (ref. 42) (arguments:–cut tail,–cut-window-size,–cut-mean-quality, -max_len1,-max_len2); (3) mapping and variant calling using bwa and IVAR v.1.2 (variant calling cut-offs: minimum quality for SNP calling = 20, minimum frequency to call a SNP = 0.05, minimum depth for calling a SNP = 20, consensus construction cut-offs: minimum quality for consensus calling = 20, minimum frequency to consider fixed a SNP = 0.8, minimum position depth = 30 (ambiguous base otherwise)) and (4) quality control assessment with MultiQC43.
Global alignment and phylogenetic reconstruction
To build the global alignment, we downloaded and concatenated all non-Spanish sequences present in GISAID11 on 21 June 2020 that passed strict filtering criteria: (1) sequences should be more than 29,000 bp in length, (2) verified insertions/deletions and (3) less than 1% of Ns and less than 0.05% of unique amino acid mutations (compared with other sequences in GISAID).
Later, we added all Spanish sequences deposited in GISAID up to 29 July 2020. The final alignment constructed included 32,914 sequences. The accession numbers of the sequences used in this study can be found in Supplementary Table 1.
Sequences were aligned against the SARS-CoV-2 reference genome44 using MAFFT45. Specific positions that have been reported to be problematic for phylogenetic reconstruction46 were masked, following the procedure described by Lanfear47, using the mask_alignment.sh script.
Finally, a maximum-likelihood (ML) phylogeny was reconstructed using IQTREE48 with the GTR model and based on the complete masked genome alignment. This phylogeny was rooted to the SARS-CoV-2 sequence obtained in Wuhan on 24 December 2019 (GISAID ID: EPI_ISL_402123).
Identification of introductions and transmission clusters
We identified transmission groups between Spanish sequences by inspecting the global phylogeny (32,914 leaves) and searching for Spanish sequences (or groups of) that were embedded within sequences with other geographic origins. Given the general low diversity among sequences, most phylogenetic nodes ended up being polytomic in the ML tree. Because of this, we defined three different transmission scenarios: (1) strains that represent introductions in Spain but differ from those from other countries and form well defined transmission groups (‘candidate transmission clusters’); (2) strains introduced into Spain that are equal to other Spanish sequences and that are also equal to sequences from other countries (‘zero-distance clusters’) and (3) Spanish sequences found within groups of sequences from other countries and which are not phylogenetically near any other Spanish sequences (‘unique’). The ‘candidate transmission clusters’ were identified as monophyletic groups of sequences composed exclusively by Spanish sequences in the phylogeny. The ‘zero-distance clusters’ were identified as Spanish sequences that share a common ancestor and that are at zero SNP distance from each other. Finally, the ‘unique’ sequences were identified as those sequences that do not share their most recent ancestor with any other Spanish sequence.
Next, we inferred how many of these transmission groups have a potential contagion date for their first case that predates the start of mobility restrictions, on 14 March 2020, by subtracting 14 days from the diagnosis date.
Finally, we wanted to investigate the international origin of these introductions. For each of the identified groups or ‘unique’ sequences with an inferred contagion date before 14 March 2020, we looked for the closest non-Spanish sequence in the phylogeny with a diagnosis date predating the first case of the transmission group. As the current consensus is that the pandemic began in Asia and later it moved to Europe, we considered only those sequences with an Asian or European origin as potential sources of introductions.
SEC alignment and phylogeny
Using the global phylogeny, we identified nodes that had at least 20 leaves and in which at least 50% of these correspond to Spanish sequences. Next, for each of these nodes or clades we reconstructed an alignment of the complete masked genomes including: (1) the sequences that belong to the identified clade; (2) 11 basal sequences from Wuhan acting as an ‘anchor’ for the phylogeny (Supplementary Table 1) and (3) a subset of 51 representative sequences, each one from a different pangolin lineage, selected to maximize the global SARS-CoV-2 genetic diversity (Supplementary Table 1, downloaded from GISAID on 20 July 2020).
For each of these alignments we inferred a ML phylogeny, using IQTREE48, with the model GTR, 1,000 fast-bootstrap replicates and rooted to the Wuhan sequence (EPI_ISL_402123). Then, in the resulting phylogeny, we identified less inclusive nodes embedded within the above identified clades and had a bootstrap support value > 80. These clades were named as potential SECs. The iTOL tool49 was used for phylogenetic visualization.
SEC8 detailed analysis
To get more detail on SEC8 phylogenetic structure, and to evaluate if mobility restrictions were effective to hinder SEC8 transmission, we enriched the original SEC8 phylogenetic tree with all the isolates of this clade sampled from February to October 2020 by the SeqCOVID consortium (959 sequences in total). Later, epidemiological information was included and plotted in the tree using the iTOL tool49.
SEC8 potential superspreading events were defined as groups of more than ten sequences, having at least one SNP in common and having a within-sequence median distance from one to three SNPs.
Population genetics and differentiation geography
Geographic distance between sequences were computed using the GPS coordinates of the patient residence city and applying the Vicenty (ellipsoid) method. Genetic diversity was calculated with two different methods: (1) genetic distance between each pair of samples in number of substitutions (SNPs), and (2) number of base substitutions per site averaged over all sequence pairs in a group of sequences. Both values have been estimated using the MEGA software50, skipping one position when a gap is found in the two compared sequences.
Demographic data for all Spanish regions and municipalities were downloaded from INE (https://www.ine.es/), and had been updated on 1 January 2020.
The genetic diversity heatmap of the Comunidad Valenciana autonomous region was generated with QGIS v.3.14.16-Pi51, using the inverse distance weighting (IDW) algorithm to interpolate the mean genetic diversity of each municipality for which we had at least two sequences.
To compare the genetic and geographic distance distribution between the different SECs, we used a one-way ANOVA test, followed by multiple pairwise-comparisons of the between-groups mean with a Tukey HSD (honestly significant difference) test.
Dating analyses
To estimate the most recent common ancestor (MRCA) of each of the nine SECs defined above, a multisequence alignment was performed including the 11 samples belonging to basal phylogenetic clades and the 51 representative sequences from different lineages (Supplementary Table 1). Before phylogenetic dating, root-to-tip regression of genetic divergence against sampling dates was performed to investigate the molecular clock signal of SECs using TempEst v.1.5.3 (ref. 52). We implemented a coalescent Bayesian exponential growth model available in Beast 2.6 (ref. 53) with the HKY + Γ model of nucleotide substitution. Tree priors were defined as follows: for effective population size we used a lognormal distribution (mean(M) = 1, standard deviation (S) = 2) and for growth rate a Laplace distribution (M = 0, S = 100). The uncorrelated lognormal relaxed clock was selected as the best fitting clock model using Bayes Factor comparisons of strict and relaxed clocks based on path sampling/stepping stone analysis54. Clock priors were defined as: ucld.mean: lognormal distribution with mean in real space = 1.4 × 10−3 subs per site per year and s.d. of the uncorrelated lognormal relaxed clock ucld.stdev = 5 × 10−2. Parameters were estimated using Markov chain Monte Carlo (MCMC) Bayesian inference, with 5 × 107 steps-long chains with exception of SEC7 and SEC8, for which longer chains were run (1 × 108); in all cases a total of 105 steps were sampled in the log files. For all analysis, three independent runs starting from different seeds were conducted to ensure convergence, then combined with LogCombiner v.2.6.3 after removing the initial 10% of the MCMC as burn-in. Adequate mixing of parameters and convergence among runs were assessed using Tracer v.1.7.1 (ref. 55) by verifying that each parameter reached an effective sampling size (ESS) above 200 and that traces showed stationarity and good mixing. The final posterior distribution contained a total of 9,000 trees, annotated with Treeannotator v.2.6.3 and visualized in FigTree v.1.4.3 (ref. 56)
Phylodynamics analysis to estimate R e
To estimate discrete changes in Re through for the two largest epidemic clades SEC7, SEC8 and two Pango lineages harboring the 614G mutation (B.1, B.1.1), we used a Bayesian birth–death skyline model (BDSKY) with serial sampling57 implemented in BEAST v.2.6 (ref. 53). BDSKY uses an episodic, piecewise birth–death model in which the parameter is allowed to change at discrete points in time, with the magnitude and timing of changes estimated from the data. In our analysis, we set two intervals wherein Re is constant and estimated the date with most evidence for a change in Re. To this end, we set a uniform prior distribution. Re was estimated before and after the changing time. The same parameters as above were used but fixing the clock rate and the recovery rate (become uninfectious rate, ẟ = 36.5 years−1) in accordance with consistent global estimates of an infectious period of 10 days58. To avoid bias in the model parameters due to constant sampling proportion assumed by BDSKY models, this parameter was set to zero before the first sample date using TreeSlicer (https://github.com/laduplessis/skylinetools/wiki). For this analysis, 1 × 108 and 4 × 108 steps-long chains were used for SEC7/B.1/B.1.1 and SEC8, respectively. Results were inspected with Tracer (v.1.7.1)55 by verifying that every parameter had effective sampling sizes above 200 and good mixing was obtained. Doubling time was calculated from the parameters estimated by BDSKY model in which growth rate (r) = (Re× δ) − δ and doubling time = ln(2)r–1.
Statistical analysis
All statistical analyses were carried out using the R statistical language59. Packages ape60, treeio60,61, doParallel62 and foreach63 were used for phylogenetic manipulation and analysis. We additionally used packages geosphere64, lwgeom65, sp66, sf67 and rgeos68 to calculate the geographic distances between samples and the geographical representation in the data. The ggplot2 R package69 was used extensively for analysis and data plotting.
Epidemic wave definitions
There is not a formal (or even official) definition for ‘first’ and ‘second’ wave, and the valley between both. We therefore used two lines of evidence to define the boundaries. One is the official number of total cases diagnosed by PCR70 and the second is the end of the mobility restrictions. On the bases of these facts, we tentatively identify the following dates for the different waves:
First wave: February 2020–14 May 2020 (from the first case reported to the end of the national lockdown and start of lifting measures).
Second wave: July 2020–first week of December 2020 (from the first large outbreaks reported after the first wave, which were caused and led to the expansion of the 20E/EU1 variant across the country to the new increase of cases in December).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the genomic sequences used in the analyses are available in the GISAID database, and the accession numbers, originating and submission laboratories can be found in Supplementary Table 1. Sequencing data (fastq files) of the samples sequenced by the SeqCOVID consortium have been deposited to the European Nucleotide Archive (ENA), and the corresponding accession numbers can also be found in Supplementary Table 1.
Code availability
The analysis pipeline used to map and analyze the sequences is available at https://gitlab.com/fisabio-ngs/sars-cov2-mapping.
References
Zhu, N. et al. Brief report: a novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727 (2020).
Spiteri, G. et al. First cases of coronavirus disease 2019 (COVID-19) in the WHO European Region, 24 January to 21 February 2020. Eurosurveillance 25, PMC7068164 (2020).
Centro de Coordinación de Alertas y Emergencias Sanitarias. Enfermedad por el coronavirus, situación en España https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Actualizacion_153_COVID-19.pdf (2020).
Pollán, M. et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): a nationwide, population-based seroepidemiological study. Lancet 396, 535–544 (2020).
Ministerio de la Presidencia, R. C. las C. y. M. D. BOE-A-2020-3692 https://www.boe.es/eli/es/rd/2020/03/14/463/con (2020).
Instituto de Salud Carlos III, Spanish Government. COVID-19 reported cases in Spain https://cnecovid.isciii.es/covid19/#ccaa (2020).
Centro de Coordinación de Alertas y Emergencias Sanitarias, Ministerio de Sanidad. Enfermedad por el coronavirus (COVID-19) https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Actualizacion_268_COVID-19.pdf (2020).
Grenfell, B. T. et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332 (2004).
Volz, E. M., Kosakovsky Pond, S. L., Ward, M. J., Leigh Brown, A. J. & Frost, S. D. W. Phylodynamics of infectious disease epidemics. Genetics 183, 1421–1430 (2009).
Vasylyeva, T. I. et al. Phylodynamics helps to evaluate the impact of an HIV prevention intervention. Viruses 12, 469 (2020).
Shu, Y. & McCauley, J. GISAID: global initiative on sharing all influenza data – from vision to reality. Euro Surveill. 22, 30494 (2017).
Callaway, E. The coronavirus is mutating – does it matter? Nature 585, 174–177 (2020).
Kupferschmidt, K. The pandemic virus is slowly mutating. But does it matter? Science 369, 238–239 (2020).
Morel, B. et al. Phylogenetic analysis of SARS-CoV-2 data is difficult. Mol. Biol. Evol. 38, 1777–1791 (2020).
Worobey, M. et al. The emergence of SARS-CoV-2 in Europe and North America. Science 370, 564–570 (2020).
Geoghegan, J. L. et al. Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand. Nat. Commun. 11, 6351 (2020).
Seemann, T. et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 11, 4376 (2020).
Alm, E. et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Eurosurveillance 25, 2001410 (2020).
Oude Munnink, B. B. et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 26, 1405–1410 (2020).
Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369, 1255–1260 (2020).
Guirao, A. The Covid-19 outbreak in Spain. A simple dynamics model, some lessons, and a theoretical framework for control response. Infect. Dis. Model. 5, 652 (2020).
Hyafil, A. & Moriña, D. Analysis of the impact of lockdown on the reproduction number of the SARS-Cov-2 in Spain. Gac. Sanit. 35, 453–458 (2021).
Lurie, M. N., Silva, J., Yorlets, R. R., Tao, J. & Chan, P. A. Coronavirus disease 2019 epidemic doubling time in the United States before and during stay-at-home restrictions. J. Infect. Dis. 222, 1601–1606 (2020).
Böhmer, M. M. et al. Investigation of a COVID-19 outbreak in Germany resulting from a single travel-associated primary case: a case series. Lancet Infect. Dis. 20, 920–928 (2020).
Rambaut, A. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 5, 1403–1407 (2020).
Lai, A. et al. Molecular tracing of SARS-CoV-2 in Italy in the first three months of the epidemic.Viruses 12, 798 (2020).
Hodcroft, E. & Neher, R. Phylogenetic analysis of SARS-CoV-2 diversity in Europe (Italy) https://nextstrain.org/groups/neherlab/ncov/italy (2021).
Plante, JA. et al. Spike mutation D614G alters SARS-CoV-2 fitness.Nature 592, 116–121 (2021).
Volz, EM. et al. Evaluating the effects of SARS-CoV-2 Spike mutation D614G on transmissibility and pathogenicity. Cell 184, 64–75.e11 (2021).
Korber, B. et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 812–827 (2020).
van Dorp, L. et al. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 11, 5986 (2020).
COVID-19 community mobility reports. Google https://www.google.com/covid19/mobility/ (2021).
Russell, TW. et al. Effect of internationally imported cases on internal spread of COVID-19: a mathematical modelling study.Lancet Public Health 6, E12–E20 (2021).
Popa, A. et al. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2.Sci. Transl. Med. 12, eabe2555 (2020).
Lemieux, J. et al. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 371, 6529 (2021).
Hodcroft, E. B. et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 595, 707–712 (2021).
Argimón, S. et al. Microreact: visualizing and sharing data for genomic epidemiology and phylogeography. Microb. Genomics 2, e000093 (2016).
Quick, J. nCoV-2019 sequencing protocol. protocols.io https://doi.org/10.17504/protocols.io.bbmuik6w (2020).
artic-network. artic-network/artic-ncov2019. github https://github.com/artic-network/artic-ncov2019 (2019).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
De Maio, N. et al. Issues with SARS-CoV-2 sequencing data. virological.org https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (2020).
Lanfear, R. A global phylogeny of SARS-CoV-2 sequences from GISAID. Zenodo https://doi.org/10.5281/ZENODO.3958883 (2020).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Kumar, S., Stecher, G., Peterson, D. & Tamura, K. MEGA-CC: computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 28, 2685–2686 (2012).
QGIS Geographic Information System (QGIS Development Team, 2020); https://qgis.org
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019).
Grummer, J. A., Bryson, R. W. & Reeder, T. W. Species delimitation using Bayes factors: simulations and application to the Sceloporus scalaris species group (Squamata: Phrynosomatidae). Syst. Biol. 63, 119–133 (2014).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Rambaut, A. FigTree http://tree.bio.ed.ac.uk/software/figtree/ (2016).
Stadler, T., Kühnert, D., Bonhoeffer, S. & Drummond, A. J. Birth–death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl Acad. Sci. USA 110, 228–233 (2013).
He, X. et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26, 672–675 (2020).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2017).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Wang, L.-G. et al. Treeio: an R package for phylogenetic tree input and output with richly annotated and associated data. Mol. Biol. Evol. 37, 599–603 (2020).
Microsoft Corporation & Weston, S. doParallel: Foreach Parallel Adaptor for the ‘Parallel’ Package (Comprehensive R Archive Network (CRAN), 2019).
Microsoft Corporation & Weston, S. foreach: Provides Foreach Looping Construct (CRAN, 2020).
Hijmans, R. J., Williams, E. & Vennes, C. Spherical Trigonometry (CRAN, 2019).
Pebesma, E., Rundel, C. & Teucher, A. lwgeom: Bindings to Selected ‘liblwgeom’ Functions for Simple Features (CRAN, 2020).
Bivand, R. S., Pebesma, E. & Gómez-Rubio, V. Applied Spatial Data Analysis with R (Springer Science & Business Media, 2013).
Pebesma, E. Simple features for R: standardized support for spatial vector data. R J. 10, 439–446 (2018).
Bivand, R. et al. rgeos: Interface to Geometry Engine – Open Source (‘GEOS’) (CRAN, 2020).
Wilkinson, L. ggplot2: Elegant graphics for data analysis by WICKHAM, H. Biometrics 67, 678–679 (2011).
Instituto de Salud Carlos III. COVID-19: Data and Documentation (Spanish Government, 2021).
Acknowledgements
This work was mainly funded by the Instituto de Salud Carlos III project COV20/00140, with additional funding by Spanish National Research Council project CSIC-COV19-021, Ministerio de Ciencia project PID2019-104477RB-100, ERC StG 638553 and ERC CoG 101001038 to I.C., and BFU2017-89594R to F.G.C. M.C. is supported by Ramón y Cajal program from Ministerio de Ciencia and grants RTI2018-094399-A-I00 and Generalitat Valenciana (Regional Government) project SEJI/2019/011. We gratefully acknowledge Hospital Universitari Vall d’Hebron, Instituto de Salud Carlos III, IrsiCaixa AIDS Research Lab and all the international researchers and institutions that submitted sequenced SARS-CoV-2 genomes to the GISAID’s EpiCov Database (Supplementary Table 1), as an important part of our analyses has been made possible by the sharing of their work. We also thank Unidad de Bioinformática y Estadística, Centro de Investigación Príncipe Felipe, for allowing us to use the Computer Cluster to perform some of the bioinformatic analysis.
Author information
Authors and Affiliations
Consortia
Contributions
I.C., F.G.C. and M.C. conceived the work. G.A.G., G.D.A. and S.J.S. set up the bioinformatics environment and the analysis pipeline. M.G.L., A.C.O., P.R.R. and N.G.G. analyzed the data. A.C.O. and M.G.L. wrote the first version of the draft. A.O., E.M., A.E.B., A.N., D.G.V., L.P.L., M.H., J.S., C.L.C., M.T., M.P.B.E., N.G.J., G.M., L.M.P., P.R.H., M.A.B., N.G.G., L.R.R., M.T.P., I.G.N., J.F.P. and A.S. sequenced genomes. G.C.E., M.M.R., L.P.V., J.M.M., R.M.M., M.D.T.B., J.A.L., V.G.G., A.R.V., D.N., E.A., I.T., A.C.B., S.H.C., M.L.C.R., A.R., A.T., A.M.B., J.M.N.M., I.P.C., S.S.G., B.F.E., C.G.C., B.P.B., I.T.P., A.C., V.M., M.P.G., L.R.F., J.L.P., J.A., J.J.C.A., M.C.P.G., J.A.B., N.R., J.L.L.H., M.A.Z. and M.P.R. provided samples. I.C., F.G.C., M.C., A.C.O., M.G.L., S.D. and D.G.V. critically reviewed and contributed to the final version of the paper. The remaining SeqCOVID-Spain consortium members provided samples, and clinical and epidemiological information.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Genetics thanks R. Rockett and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Abundance of the different Pango lineages in the dataset.
In the x-axis, the epidemiological week as plotted in Microreact.
Extended Data Fig. 2 Examples of the different groups of sequences identified.
‘Candidate transmission clusters’ are groups of Spanish sequences that form a clade. ‘Zero distance clusters’ are groups of Spanish sequences that are at zero distance from each other. Finally, the ‘unique’ sequences are Spanish sequences that are more than 1 SNP away from any other Spanish sequence and that do not share a most recent common ancestor (MRCA) node with other Spanish sequences.
Extended Data Fig. 3
Number of international and Spanish sequences in each SEC.
Extended Data Fig. 4 Phylogenetic location of each SEC in the global SARS-CoV-2 phylogeny.
Sequences from Spain are colored according to their SEC color (as indicated in Fig. 2a legend) while international sequences remain in black color.
Extended Data Fig. 5 Distribution of genetic (salmon) versus geographic (gray) distances within each pair of samples belonging to the same SEC.
For each SEC we had the following comparisons (data points): SEC1 (N = 120), SEC2 (N = 190), SEC3 (N = 91), SEC4 (N = 325), SEC5 (N = 378), SEC6 (N = 990), SEC7 (N = 14,028), SEC8 (N = 178,503) and SEC9 (N = 231). The lower whisker, higher whisker, center and bounds of each boxplot refers to quartile 1–1.5 interquartile range, quartile 3 + 1.5 interquartile range, mean, first and third quartiles of the data. Individual points are outliers (values lower than quartile 1–1.5 interquartile range and higher than quartile 3 + 1.5 interquartile range).
Extended Data Fig. 6 Distribution of sequences harboring the 614 G mutation (blue) versus the 614D mutation (salmon,wild-type) in the S gene for the spanish sequences in our dataset.
In the left panel, a histogram of samples sorted by date of sequencing. At right, frequency of both mutations in the sequenced samples by date. The national lockdown event is marked by a purple vertical line. At the bottom, ‘candidate transmission clusters’ by date and size, colored according to the allele found at the 614 position of the S gene.
Extended Data Fig. 7 Cases sequenced during the period that includes the first wave until the end of the lockdown (14th May, 2020).
Lines represent the number of cases belonging to different Spanish Epidemic Clades (SECs).
Extended Data Fig. 8 Phylodynamic estimates of the effective reproductive number (Re) of Spanish SEC7.
A birth–death skyline (BDSKY) model was implemented in Beast v.2, allowing for piecewise changes in Re, with the time and magnitude estimated from the data. The X axis represents time, starting with the MRCA of all sampled diversity within SEC7 and ending with the date of the most recently sequenced genome from 15th May. The blue dotted line indicates the posterior value of the timing of a most important decrease in Re, around 20th March 2020 [95% HPD: 15–25th March]. The Y axis represents Re, and the violin plots show the posterior probability distribution for this parameter before and after the change time in Re; with a mean of 2.10 [95% HPD: 1.67–2.62] and 0.27 [95% HPD:0.06–0.47] before and after this time, respectively. The phylogenetic tree in the background is the maximum clade credibility tree from the BDSKY analysis, with the tips colored according to whether they were sampled before or after 20th March. The lower whisker, higher whisker, center and bounds of each boxplot refers to quartile 1–1.5 interquartile range, quartile 3 + 1.5 interquartile range, mean, first and third quartiles of the data. Individual points are outliers (values lower than quartile 1–1.5 interquartile range and higher than quartile 3 + 1.5 interquartile range). Boxplot was constructed with all the Spanish sequences in SEC7 (N = 182).
Extended Data Fig. 9 Comparison between strains carrying 614D and 614 G mutations.
a. Number of sequences belonging to SEC7, SEC8 (614D) and those having the 614 G allele. b. Phylodynamic estimates of the effective reproductive number (Re) of clades harboring 614D mutation (SEC7 and SEC8) and Pango lineages with 614 G mutation (B.1 and B1.1). A birth–death skyline (BDSKY) model was implemented in Beast v.2, allowing for piecewise changes in Re, with the time and magnitude estimated from the data. The violin plots show the posterior probability distribution (HPD) interval for Re parameter before (Re1) and after (Re2) the changing time estimates (dotted line). For SEC7, Re1: 2.10 [95% HPD: 1.67–2.62] and Re2: 0.27 [95% HPD:0.06–0.47]; changing time 20th March 2020 [95% HPD: 15–25th March]. For SEC8, Re1: 3.14 [95% HPD: 2.71–3.57] and Re2: 0.23 [95% HPD: 0.15–0.32]; changing time 9th March 2020 [95% HPD: 8–10th March]. For B.1, Re1: 1.86 [1.6–2.16] and Re2: 0.26 [0.14–0.41]; changing time 23rd March 2020 [95% HPD: 21–25th March]. For B.1.1, Re1: 1.62 [1.44–1.80] and Re2: 0.15 [0.05–0.29]; changing time 10th April 2020 [95% HPD: 9–12th April]. The lower whisker, higher whisker, center and bounds of each boxplot refers to quartile 1–1.5 interquartile range, quartile 3 + 1.5 interquartile range, mean, first and third quartiles of the data. Individual points are outliers (values lower than quartile 1–1.5 interquartile range and higher than quartile 3 + 1.5 interquartile range). Boxplots were constructed with all the Spanish sequences in SEC7 (N = 182), SEC8 (N = 636), B.1 (N = 191) and B.1.1 (N = 223).
Supplementary information
Supplementary Information
Supplementary Notes and Figs. 1–6.
Supplementary Data 1
Alignment and phylogenetic tree of the Spanish sequenced samples, to be plotted using the Microreact server.
Supplementary Video 1
Video showing the transmission dynamics of SEC8 within Spain from 2020-01-16 to 2020-07-19, obtained from NextStrain.
Supplementary Tables
Supplementary Tables 1–3.
Rights and permissions
About this article

Cite this article
López, M.G., Chiner-Oms, Á., García de Viedma, D. et al. The first wave of the COVID-19 epidemic in Spain was associated with early introductions and fast spread of a dominating genetic variant. Nat Genet 53, 1405–1414 (2021). https://doi.org/10.1038/s41588-021-00936-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00936-6
This article is cited by
-
Dental injury offenses and compensation imposed by Spanish courts, before and during Covid-19. A cross-sectional study
Scientific Reports (2023)
-
The molecular epidemiology of SARS-CoV-2 in the Pityusic Islands shows multiple introductions and fast replacements of variants in a touristic worldwide hot spot
Scientific Reports (2023)
-
Global landscape of SARS-CoV-2 genomic surveillance and data sharing
Nature Genetics (2022)
