
Abstract
As the SARS-CoV-2 virus spreads through human populations, the unprecedented accumulation of viral genome sequences is ushering in a new era of ‘genomic contact tracing’—that is, using viral genomes to trace local transmission dynamics. However, because the viral phylogeny is already so large—and will undoubtedly grow many fold—placing new sequences onto the tree has emerged as a barrier to real-time genomic contact tracing. Here, we resolve this challenge by building an efficient tree-based data structure encoding the inferred evolutionary history of the virus. We demonstrate that our approach greatly improves the speed of phylogenetic placement of new samples and data visualization, making it possible to complete the placements under the constraints of real-time contact tracing. Thus, our method addresses an important need for maintaining a fully updated reference phylogeny. We make these tools available to the research community through the University of California Santa Cruz SARS-CoV-2 Genome Browser to enable rapid cross-referencing of information in new virus sequences with an ever-expanding array of molecular and structural biology data. The methods described here will empower research and genomic contact tracing for SARS-CoV-2 specifically for laboratories worldwide.
Similar content being viewed by others

Main
In the past year, the SARS-CoV-2 virus quickly spread across human populations worldwide1,2,3. Recent technological advances have enabled rapid and cost-efficient sequencing of the viral genome—over 2,000 groups worldwide have generated 97,733 high coverage whole-genome SARS-CoV-2 sequences that are available on GISAID4 as of 24 September 2020. These vast datasets and rapid sequencing turnaround times are enabling a type of ‘genomic contact tracing’ where genetic similarities between viral genomes isolated for different hosts carry important information about the transmission dynamics of the virus. For example, these data can be used to infer the number of unique introductions of the viral genome in a given area5,6,7,8,9,10,11 and identify ‘transmission chains’ among seemingly unrelated infections12,13,14,15,16.
Despite great potential, this unprecedented and ongoing accumulation of sequencing data is overwhelming existing systems for the analysis and interpretation of viral transmission and evolutionary dynamics. In part, this is because typical phylogenetic applications accumulate all of the relevant sequence data before beginning phylogenetic inference. For genomic contact tracing to work effectively, each new viral genome sequence must be contextualized within the entire evolutionary history of the virus rapidly and accurately as it is collected. This could be accomplished by re-inferring the full phylogeny, but with current SARS-CoV-2 datasets containing hundreds of thousands of samples, this takes more than a day even using powerful computational resources. Alternatively, new genome sequences could be contextualized by placing samples onto an existing ‘reference phylogeny’ and several methods have been developed for this purpose17,18,19,20. These methods have been used to place new samples onto a phylogeny created from a small subset of available SARS-CoV-2 isolates21 and provide regular updates to a global phylogeny of SARS-CoV-2 (ref. 22). Nonetheless, existing algorithms for placing sequences onto reference phylogenies are far too slow to enable real-time genomic contact tracing (Table 1).
Quantification of uncertainty is a fundamental aspect of interpreting phylogenetic inferences23 and sample placements onto a reference phylogeny. Nonparametric bootstrapping24 has been a cornerstone of phylogenetic inference for decades but this is impractical for the extremely large sample sizes and limited phylogenetic information in SARS-CoV-2 genome isolates. More recently developed methods, such as ultrafast bootstrapping25,26, are fast but not applicable to the problem of placing individual samples onto a reference phylogeny. An alternative to these approaches is the approximate likelihood-ratio test27 but its computation is prohibitively slow and interpretation challenging. Therefore, quantifying uncertainty in sample placement on reference phylogenies is an important unsolved problem and particularly relevant during this pandemic.
In this work, we describe an efficient method that facilitates rapid, maximum parsimony addition of samples onto an existing phylogeny. We show that our method for placing new genome sequences onto a SARS-CoV-2 phylogeny is orders of magnitude faster than existing approaches and produces highly accurate results. Additionally, we introduce the branch parsimony score (BPS), which is the minimum number of additional mutations (the parsimony score) required to accommodate a sample placement at a given branch. This offers an intuitive means of quantifying uncertainty in sample placements for SARS-CoV-2 phylogenies. Our placement approach and related data visualization tools are available from the University of California Santa Cruz (UCSC) SARS-CoV-2 Genome Browser28 and will empower genomic contact tracing applications worldwide.
Results
Prior tools are inadequate for SARS-CoV-2 phylogenetics
Genomic contact tracing during this global pandemic necessitates algorithms that efficiently place samples onto the vast global tree. With this requirement in mind, we evaluated the performance of several existing approaches17,18,19,20 and compared their runtime and memory usage by adding just 1 additional sequence to a SARS-CoV-2 global phylogeny containing 38,342 leaves, our ‘reference phylogeny’, which comes from the 11 July 2020 release of Robert Lanfear’s global SARS-CoV-2 phylogeny22. We found that the time and memory required to place a single sample is unacceptably large. For example, EPA-ng18 takes approximately 28 central processing unit (CPU) minutes to place one sample and requires 791 gigabytes (GB) of memory (Table 1).
To address the challenge of real-time sample placement, we developed a new tool called Ultrafast Sample placement on Existing tRees (UShER). UShER can place a SARS-CoV-2 sample onto our reference phylogeny in just 0.5 s, which is a 3–4 orders of magnitude improvement over the next fastest tool (Table 1). A part of the increased efficiency of UShER stems from its heavily optimized encoding of mutations compared to a multiple sequence alignment (MSA) and from its precomputed data object storing the inferred histories of mutation events on the tree before placing samples during each execution.
UShER uses efficient tree-based data objects
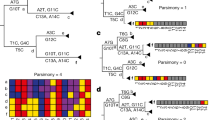
Existing approaches to sample placement use an MSA of genomes that requires storing a whole-genome sequence for each sample (Fig. 1 and Methods). UShER’s primary data structure is substantially more efficient. It starts with a list of variants with respect to a reference sequence for each sample and represents genotype data based on the inferred phylogeny of the viral population itself. UShER uses the Fitch–Sankoff algorithm to infer the placement of mutations on a given tree and on the variant list29,30. Besides the phylogeny itself, UShER records only the nodes for which mutations are inferred to have occurred on the branches leading to them in a representation that we call mutation-annotated tree (Fig. 1). This representation is particularly favorable for the SARS-CoV-2 phylogeny where the mutations are relatively rare and often shared across several samples. This approach has parallels to efficient tree-based representations used recently in population genetics31,32. For our SARS-CoV-2 reference phylogeny, UShER’s mutation-annotated tree uses only 3.4 megabytes (MB) of memory, which fits easily in a last-level cache33, to encode virtually the same information as the full MSA, which requires 1.14 GB (>300× improvement).

a, Prior methods rely on a full MSA to inform phylogenetic structure (left), while UShER uses a mutation-annotated tree (right). The MSA shown is color-coded to match the mutations present in the tree above (A, red; C, yellow; G, purple; U, blue). b, UShER evaluations of the parsimony score for placing the sample S5 (blue) at each possible position (Methods) of our example phylogeny (shown in a). We considered the branch leading to a given node to be the parent branch. The branches that need to be modified or added to the phylogeny to accommodate S5 are shown in blue; back mutations (if present) are colored red and new nodes are circled. For example, if S5 is placed at S1, the new node 3 has children S1 and S5 and two back mutations (U4C and A6G) occur at the branch leading to S5, giving this placement a parsimony score of 2. Placing S5 at node 1 is optimal by parsimony. c, The final tree with S5 added, where an additional internal node 3 is added to support S5 (left); the mutation annotations for the final tree with S5 colored in blue are shown on the right. Note that the memory efficiency of the mutation-annotated tree can vary depending on the dataset. In all panels, the length of each branch is proportional to the number of mutations that occurred on that branch. Zero-length branches, which are not associated with any mutations (for example, those leading to node 3 in ‘at root’, ‘at S1’, ‘at S2’, ‘at S3’ and ‘at S4’ in b) are shown as very short branches for visibility.
UShER can generate a mutation-annotated tree for our reference tree with 38,342 leaves and 15,129 variant positions in just 2 min 24 s using 4 threads (Supplementary Table 1). This data structure is then stored as a preprocessed protocol buffer (https://developers.google.com/protocol-buffers), which is a customizable binary file that can be rapidly loaded (approximately 150 ms) during sample placement and data visualization (Fig. 1) and obviates the need to recompute the assignments for each execution.
Sample placement using a mutation-annotated tree
UShER uses this mutation-annotated tree to rapidly place newly acquired samples onto the tree of SARS-CoV-2 variation. More specifically, UShER uses a maximum parsimony approach where it searches the entire reference tree (Fig. 1 and Methods) for a placement that requires the fewest additional mutations to accommodate the added sample (that is, the maximum parsimony placement of a sample). UShER breaks ties based on the number of descendant leaves at the placement nodes when multiple placements are parsimony-optimal (Methods). When a preprocessed mutation-annotated tree is already available, this procedure takes approximately 0.5 s to place a single sample onto the SARS-CoV-2 reference tree (Table 1) and is even more efficient when placing larger sets of samples since the time to load the mutation-annotated tree is gradually reduced. For example, it only takes approximately 18 s to place 1,000 samples onto our reference tree using 16 threads (Supplementary Table 1). This means that our implementation is fast enough to facilitate real-time placement of SARS-CoV-2 sequences and is sufficiently memory efficient (Table 1 and Supplementary Tables 1 and 2) that everything we present could be run on a basic laptop; this should facilitate widespread adoption of this approach.
UShER accurately places simulated SARS-CoV-2 samples
To evaluate the accuracy of UShER’s maximum parsimony-based placement algorithm when the viral evolutionary history is known, we generated a SARS-CoV-2-simulated dataset using a fixed tree that we supplied (Methods). UShER places samples with the correct sister node in 97.2% of cases. For samples with just 1 parsimony-optimal placement, UShER achieves 98.5% accurate local placements. When incorrect, UShER’s placements tend to be quite close to the correct node on the SARS-CoV-2 global phylogeny, that is, separated by just 1.1 edges from the correct position on the tree on average (Fig. 2 and Methods). Therefore, we conclude that UShER is capable of accurately placing new samples onto a fixed SARS-CoV-2 global reference phylogeny in practice. Although UShER works well for SARS-CoV-2, it will not be as accurate for phylogenetic analyses where maximum parsimony algorithms are known to perform poorly (for example, cases of long branch attraction34).

Top: We independently masked sites at 10, 20, 30, 40 and 50 percent of sites for each of 10 simulated genomes to be added to the phylogeny and computed the Robinson–Foulds distance51, the average number of lineages added that had identical sister node sets to those in the simulated reference tree, the distance from true placement for each lineage added (Methods) and the number of equally parsimonious sites per placement for each lineage added. Middle: We introduced random nucleotide substitutions to the genomes of the 10 lineages added to the tree by UShER at a rate of 1, 2, … 10 sites per genome, drawn independently, and computed the same measures of coherence to the reference tree, with the error bars representing the 95% confidence intervals. Bottom: We introduced one systematic error to 1, 2, … 10 of the genomes added to the tree by UShER and computed the same metrics as above. For each experiment, the distance from true placement was strongly correlated with the amount of missing data (P < 3.34 × 10−112 for all experiments; Spearman rank correlation test with 5,998, 10,998 and 10,998 d.f. for the masking, random error and systematic error experiments, respectively). For each panel depicting Robinson–Foulds scores, the distribution of scores across 100 replicates where 10 lineages were added randomly to the phylogeny is shown to the far right for a null model comparison and is labeled ‘Null’. n = 100 replicates for each experiment. Each box plot is centered on the median of the data and extends to the first and third quartiles, with the lower whiskers extending to the lowest data point within the first quartile minus 1.5 times the interquartile range and the upper whiskers extending to the highest data point within the third quartile plus 1.5 times the interquartile range. In the error bar panels (second from the left), the data points are centered on the mean of the data and extend to the bounds of the 95% confidence interval, calculated by 1,000 iterations of bootstrapping.
Missing data affect placement of SARS-CoV-2 genomes
Given the low mutation rate and therefore low phylogenetic signal in SARS-CoV-2 viral genomes, missing data have a large impact on phylogenetic placement, as expected (Fig. 2). When we randomly masked between 0 and 50% of positions in samples to be placed by UShER, all measures of placement accuracy were negatively impacted. With 50% of all sites masked, only 41.9% of samples were assigned identical sister nodes as their true placement on the reference tree. However, the mean distance between UShER and correct placements on the tree remained relatively small—just 1.61 edges—and 81.0% of lineages had sister node sets in the UShER tree that were a subset of the sister nodes in the reference tree or vice versa (Methods).
High rates of missing data have a slightly larger effect on the precision of UShER’s placements than for maximum-likelihood tree inference methods when constructing a complete subtree de novo (Extended Data Fig. 1). When using Robinson–Foulds distance to measure congruence with the correct tree, we found that when no sites were masked, the average distance values from the correct tree for the trees obtained from the three methods were within 12.7% of each other and 12.9–13.3 times lower than a null model obtained from random tree construction (Extended Data Fig. 1). With no missing data, UShER produces the most congruent tree (that is, having the lowest Robinson–Foulds distance) to the correct tree, on average. The distance values increased by up to 11.1% with only 2.5% missing data and up to 76% with 10% missing data, with UShER being slightly more adversely affected by missing data than the other methods. Based on these observations, we recommend that the reference tree should ideally be maintained using only genomes with nearly complete sequences regardless of the tree inference method (for example, by filtering data obtained from the GISAID database using ‘complete’ and ‘high coverage’ tags).
UShER is robust to low error rates in SARS-CoV-2 genomes
Two types of errors in SARS-CoV-2 consensus sequences also affect the accuracy of sample placements. First, stochastic errors are likely present in many available SARS-CoV-2 sequences35. When we simulated independent errors, we found that the effects on UShER’s accuracy were modest (Fig. 2). With 10 errors on average, placement is approximately 20% less likely to select the correct sister node; other distance metrics are similarly impacted (Fig. 2). Our results indicate that especially low-quality samples should be rigorously identified and excluded from analyses using UShER. Additionally, poor-quality samples can be easily flagged because they will tend to appear as unrealistically long terminal branches in UShER’s placements. UShER reports all newly added samples with a parsimony score >3 along with a list of parsimony-increasing sites.
Second, systematic errors, where the same apparent variant is introduced into many sequences, are present in some SARS-CoV-2 sequences and have the potential to affect phylogenetic inference because they appear as inherited mutations36,37. Whereas UShER appears to be robust to a single systematic error present in fewer than 5 samples (Fig. 2), a single systematic error present in all 10 samples had a similar overall effect on placement accuracy as 50% missing data in error-free sequences. Consistent with our previous work36,37, the addition of two perfectly correlated systematic errors can drastically affect UShER performance (Extended Data Fig. 2). Systematic errors should be rigorously identified and removed before sample placements are performed. We refer readers to methods that we developed previously to detect and eliminate such errors36,37; the UShER package includes a tool to remove known problematic positions when preparing input data.
We emphasize that sequencing errors pose similar challenges for other phylogenetic inference tools (Extended Data Fig. 1) and our analysis is meant to serve as a guideline to the user rather than highlight the limitations of UShER.
Quantifying uncertainty in sample placement
Quantifying uncertainty in phylogenetic placement is critical for accurately interpreting SARS-CoV-2 phylogenies where the true phylogenetic signal is limited and sometimes even contradictory35,36. We developed functionality within UShER to report the number of equally parsimonious placements by default. Additionally, UShER can output the minimum number of additional mutations required to accommodate a single sample placed on each branch of the reference tree, a measure that we call the BPS. We limited this function to single sample placements because it would be challenging to quantify and represent the uncertainty imposed by the sequential incorporation of additional samples. As would be expected given the typically unambiguous sample placements for high-quality sequences on the global phylogeny, BPS typically increases rapidly with increasing distance along the tree (Fig. 3). Additionally, users can easily accommodate uncertainty in the input phylogeny by adding samples to many input trees in parallel. UShER can also quantify phylogenetic uncertainty within a set of samples to be placed by producing all possible topologies resulting from equally parsimonious sample placements (Extended Data Fig. 3 and Methods). To our knowledge, UShER is the only phylogenetic placement tool with this ability to produce all possible parsimony-optimal topologies and can achieve this because of its efficient data structures and high speed.

The correct sample placement, which corresponds to the maximally parsimonious placement, is shown by the arrow and each branch is colored by the BPS for that sample on that branch. The phylogeny shown has been randomly subsampled to include only 250 samples for clarity of presentation. Branch lengths are measured in substitutions per genome. For the purposes of generating this illustrative figure, we placed only a single randomly selected sample (n = 1).
UShER is congruous with standard methods on SARS-CoV-2 data
To evaluate the performance of our approach under realistic conditions with real world SARS-CoV-2 data, we used UShER to place real samples onto a global reference phylogeny. Because the phylogeny was necessarily inferred from real data (Methods), this approach measures the consistency of placement between more typical tree-building approaches and the UShER placement algorithm rather than placement accuracy per se. To evaluate consistency, we randomly pruned and replaced 100 sets of 10 samples each using the reference tree (Methods). We found that UShER placed each with an identical sister node as in the reference tree in 90.0% of cases (Fig. 4). Additionally, placements tended to be quite close to correct and the mean number of edges between the reference position and UShER’s placement was just 0.159 and the mean Robinson–Foulds distance for trees with 10 samples added was 1.27 (Fig. 4a–c). When we mimicked a plausible use case by removing larger sets of related sequences, we found that UShER can also accurately reconstruct larger subtrees for the added samples (Fig. 4d–g). Collectively, our metrics are not far from those we obtained when analyzing the simulated datasets; they indicate that missing data, errors and other features of real sequences occasionally impact UShER’s placements.

a–c, Robinson–Foulds distance between 100 reference and UShER-generated trees produced by removing and re-adding 10 samples in each (a), distance from the reference placement for each of 1,000 placed samples (b) and number of equally parsimonious placements for each of the 1,000 placed samples (c) are shown. d–g, Comparisons of subsets of the global phylogeny released on 11 July 2020 with reconstruction of this phylogeny using UShER. In each case, we pruned lineages colored in red from the phylogeny and added them back using UShER. UShER accurately placed randomly selected subtrees containing lineages collected in the western United States in March and April (d) and in Europe in March (e), as well as more distantly related lineages whose times and places of collection differed more widely (f,g). d, Differences in tree topology are highlighted in bold. e–g, Other topologies are identical. All trees in this figure are ultrametric and branch lengths are arbitrary.
We found that samples causing inconsistent placements between the reference tree and UShER were the mostly challenging cases. In particular, 6 of the 1,000 sequences that we attempted to place using UShER had large numbers of equally parsimonious placements (5–65) and were placed inconsistently relative to the reference tree. Each of these consensus sequences had a large number of ambiguous nucleotide positions (8–15) that overlapped many phylogenetically informative sites in the reference tree. This may suggest a mixture of two genetically divergent samples—either a true mixed infection or laboratory-induced. Regardless of the source, we believe future versions of the reference tree should rigorously filter sequences containing ambiguities at phylogenetically informative positions.
Additionally increasing genetic distance and sequencing errors are expected to affect placement accuracy. We found that samples are more likely to be placed inconsistently when the parsimony score is higher (P = 2.98 × 10−5, one-tailed Mann–Whitney U-test). Incorrectly placed samples also had significantly more equally parsimonious placements (P = 1.3 × 10−21, one-tailed Mann–Whitney U-test). In fact, 15% of real samples had more than 1 equally parsimonious placement on the reference phylogeny and many distinct nodes were identical in the reference tree. However, if we restricted the analysis to samples with only a single most parsimonious placement, we found that 97% of UShER’s placements were consistent with the maximum-likelihood reference tree. We suggest that placement of samples that are unusually genetically distant or that have many equally parsimonious placements on a reference tree should be regarded with caution. Both statistics are reported by UShER.
UShER can maintain a global phylogeny
We propose that UShER could form the basis for ‘real-time’ phylogenetic platforms in periodically updating the reference tree itself or be used in conjunction with maximum-likelihood updates. To investigate this, we used UShER to add all of the 9,437 additional sequences in the 31 July 2020 release of the global tree to our 11 July 2020 reference tree. We also extensively optimized both trees using a maximum-likelihood approach in FastTree 2 (ref. 38) (Methods and Supplementary Data 1). The Robinson–Foulds distance between all trees was similar, suggesting that the UShER updated topologies were close to de novo phylogenies (Supplementary Table 3). Additionally, the optimized version of the phylogeny produced by UShER resulted in a substantially increased likelihood over the 31 July 2020 tree inferred de novo with similarly extensive optimization (Supplementary Table 4). We obtained the highest likelihood topology from a heavily optimized 11 July 2020 tree, sample addition with UShER and then another round of tree optimization (Supplementary Table 4). This indicates that UShER, combined with additional rounds of optimization, does not result in unrecoverable local minima but rather may help avoid them. In combination with periodic maximum-likelihood updates to the global phylogeny, UShER can offer an appealing combination of real-time phylogenetic methods and model-based practices. This combination can be used to maintain an updated phylogeny for the SARS-CoV-2 pandemic.
Web interface of UShER on the UCSC SARS-CoV-2 Genome Browser
Interpretation of UShER’s placements often involves scrutinizing the relationships and genotypes among closely related samples already present in the reference tree. In addition to providing the complete phylogenetic tree with new samples added, UShER can optionally provide local subtree outputs of a specified size (number of sample leaf nodes) so that the relationship of added samples to their nearest neighbors can be visualized and examined in detail. If all added samples fit within the specified size, then one subtree is created; otherwise, multiple subtrees are created as necessary to provide local subtrees for all samples.
To make genomic contact tracing using UShER widely available, we developed a Web interface integrated with the UCSC SARS-CoV-2 Genome Browser (https://genome.ucsc.edu/cgi-bin/hgPhyloPlace). Users may upload new sample sequences in a FASTA file, or alternatively, new sample variants relative to the reference sequence (NC_045512.2/MN908947.3/Wuhan-Hu-1) in a variant call format (VCF) file. The Web server runs UShER on the new sequences and presents a summary of the sample placements to the user, with a link to download the phylogenetic tree including the newly placed sample(s). The user can click a button to view custom tracks in the Genome Browser that show subtree(s) with a configurable number of (default 50) leaves including the new sample(s) and related sequences from the initial phylogenetic tree (Extended Data Fig. 4). The Web server uses UShER’s mutation-annotated tree to provide almost instant visualization. Additionally, to facilitate tree exploration and cross-referencing sequences against privately maintained personal health information, our Web interface also generates a JSON file in the Auspice v.2 format39 for each subtree and adds a button to view each subtree using the Nextstrain’s interactive display40 (Extended Data Fig. 5). JSON files can also be downloaded and viewed behind a firewall using Auspice, which provides a drag and drop interface to view locally stored, private sample metadata (https://auspice.us40,41).
Widespread use of genomic contact tracing has the potential to enable public health practitioners to link apparently independent incidences of infection even across disparate sequencing centers, as well as disproving false links inferred from circumstantial evidence. This provides important and actionable information for suppressing transmission and refining public health practices. User-uploaded viral genome sequences are discarded shortly after use and not shared or stored on the UCSC Genome Browser servers unless the user saves the subtree custom tracks in a Genome Browser session. However, we stress that for global contact tracing to be maximally effective, most users must upload their viral genome sequences to public sequence repositories so that their data can also be incorporated into the reference tree. Therefore, we echo the call from the International Nucleotide Sequence Database Collaboration (https://ncbiinsights.ncbi.nlm.nih.gov/2020/08/17/insdc-covid-data-sharing/) for all SARS-CoV-2 sequencing datasets to be made publicly available as soon as is practical.
Discussion
The SARS-CoV-2 pandemic has been accompanied by unprecedented levels of pathogen genomic sequencing, which has given rise to the opportunity for near real-time monitoring of viral transmission and evolution. This seemingly endless flood of genome sequence data has also pushed phylogenetic analysis frameworks to the extreme of their capabilities, requiring new approaches to rapidly incorporate and contextualize newly sequenced viral genomes. UShER is an extremely efficient software package inspired by a need to study the ongoing evolution of the virus itself, which provides a method to immediately incorporate viral genome isolates into a global phylogenetic tree. Compared to its closest counterpart, UShER is over 3,000 times faster, orders of magnitude more memory efficient and enables real-time genomic contact tracing. UShER is also available to the worldwide research community through a user-friendly Web interface in the SARS-CoV-2 UCSC Genome Browser. Although several challenges still remain for routinely deploying pathogen surveillance methodologies including rapid genomic data production and sharing, and outreach for public health officers, UShER removes a key barrier to pathogen surveillance by greatly decreasing the turnaround time from sample to analysis and empowering real-time genomic contact tracing efforts during the SARS-CoV-2 pandemic and beyond.
Methods
Implementation and optimization of algorithms in UShER
Given the existing samples, whose genotypes and phylogenetic tree are known, and the genotypes of new samples, UShER aims to incorporate new samples into the phylogenetic tree while preserving the topology of existing samples and maximizing parsimony. UShER’s algorithm consists of two phases: (1) the preprocessing phase; and (2) the placement phase.
In the preprocessing phase, UShER accepts the phylogenetic tree of existing samples in a Newick format and their genotypes, specified as a set of single-nucleotide variants with respect to a reference sequence (UShER currently ignores indels), in a VCF format. For each site in the VCF, UShER uses the Fitch–Sankoff algorithm29,30 to find the most parsimonious nucleotide assignment for every node of the tree. When a sample contains ambiguous genotypes, multiple nucleotides may be most parsimonious at a node. To resolve these, UShER assigns it any one of the most parsimonious nucleotides with preference, when possible, given to the reference base. UShER also allows the VCF to specify ambiguous bases in samples using the International Union of Pure and Applied Chemistry format (https://www.bioinformatics.org/sms/iupac.html), which are also resolved to a unique base using the above strategy after inferring the most parsimonious nucleotide(s). When a branch leading to a node is found to carry a mutation, that is, the base assigned to the node differs from its parent, the mutation (for example, G6A at node 1; Fig. 1a) is added to a list of mutations corresponding to the branches leading to that node. Finally, UShER uses protocol buffers to store in a file the Newick string corresponding to the input tree and a list of lists of node mutations, which we also refer to as mutation-annotated tree object, as shown in Fig. 1. The outer list is ordered according to the depth-first traversal of nodes. UShER also parallelizes the independent Fitch–Sankoff computations for multiple VCF sites efficiently using multiple threads (Supplementary Tables 1 and 2).
In the placement phase, UShER loads the preprocessed mutation-annotated tree and the genotypes of new samples in a VCF format and sequentially adds the new samples to the tree. For each new sample, UShER computes the additional parsimony score required for placing it at every node in the current tree while considering the full path of mutations on the branches from the root of the tree to that node (Fig. 1b). For internal nodes, the parsimonious placement can be as a sibling to the node (for example, node 1 in Fig. 1b), when there are mutations on the branch leading to that node that are not shared by the new sample or as a child to that node (for example, node 2 in Fig. 1b), when all mutations on the branch leading to that node are shared by the new sample. For leaf nodes, only sibling placement is considered (Extended Data Figs. 1–4 and Fig. 1b) to ensure that samples are always maintained as leaves of the tree. Next, UShER places the new sample at the node that results in the smallest additional parsimony score. While placing a new sample, UShER parallelizes the parsimony score computation over different nodes of the tree using multiple threads.
When multiple node placements are equally parsimonious, UShER uses the following tie-breaking strategy that has good empirical results (based on simulated data). When comparing two independent equally parsimonious placements, UShER picks the node with a greater number of descendant leaves for placement. However, if the choice is between a parent and its child node, UShER picks the parent node if the number of descendant leaves of the parent that are not shared with the child node exceed the number of descendant leaves of the child. We note that maximum likelihood might perform better in tie-breaking between otherwise equivalent placements in some applications; however, the SARS-CoV-2 maximum-likelihood approaches appear to perform similarly to UShER’s strategy described above, suggesting that this is not a key challenge for our application (Extended Data Fig. 1). To limit the uncertainty in sample placement that often arises out of incomplete sequences, UShER also provides an option to specify the maximum number of equally parsimonious placements allowed for placing a sample. The default value is set high such that a placement is always carried out by default.
At the end of the placement phase, UShER allows the user to create another protocol buffer file containing the mutation-annotated tree object for the newly generated tree including added samples (Fig. 1c). This allows another round of placements to be carried out over and above the newly added samples. While UShER’s sequential placement of new samples helps it achieve high speed, the placements could potentially be worse than a full de novo tree inference procedure. However, in practice, we have found UShER’s accuracy over iterated placements to be reasonably high.
UShER implements a few additional optimizations to speed up the placement phase. For example, the parsimony score for a node requires computing the symmetric set difference between the set of new sample variants and the set of mutations on the branches from the root of the tree to that node. UShER maintains mutations sorted by positions to speed up this computation. UShER also maintains the minimum parsimony score of previously traversed nodes in a shared variable and terminates the computation of the set difference in a new node as soon as the parsimony score corresponding to it exceeds the value of this shared variable. Finally, UShER also allows an option during the preprocessing phase to collapse nodes (that is, delete the node after moving its child nodes to its parent node), branches leading to which are not inferred to contain a mutation through the Fitch–Sankoff algorithm as well as condensing nodes in a polytomy that contains identical sequences into a single representative node, both of which help in greatly reducing the search space for the placement phase.
Branch lengths in UShER
Since UShER is based on maximum parsimony, by default it reports branch length as the number of mutations assigned to that branch. However, UShER also provides an option to the user to retain the original branch lengths from the input tree, which could be, for example, maximum-likelihood estimates for substitutions per site, for all branches that are unaffected by sample placement, while branch lengths for new branches and those modified during the placement are undefined (that is, they do not have an associated value in the Newick format). We do this to avoid inconsistencies between branch lengths in the resulting augmented trees.
Enumerating all possible parsimony-optimal placement topologies in UShER
To further aid the user to quantify phylogenetic uncertainty in placement, UShER provides an option to enumerate all possible topologies resulting from equally parsimonious sample placements. UShER does this by maintaining a list of mutation-annotated trees, starting with a single mutation-annotated tree corresponding to the input tree of the existing samples, and sequentially adds new samples to each tree in the list while increasing the size of the list as needed to accommodate multiple equally parsimonious placements for a new sample. To keep the runtime and memory usage of UShER reasonable, the user specifies the maximum number of topologies that UShER should maintain before it reverts back to using the default tie-breaking strategy described above to pick a single placement among multiple equally parsimonious placements for the subsequent samples, therefore avoiding a further increase of the list size. Note that if the number of equally parsimonious placements for the initial samples is large, the tree space can get large too quickly and slow down the placement for the subsequent samples. Therefore, UShER also provides an option to sort the samples first based on the number of equally parsimonious placements.
Simulating realistic SARS-CoV-2 genome evolution
To evaluate the accuracy of our placement algorithm on a known phylogeny, we produced a set of simulated samples for which we knew the correct placement on the tree. To do this, we first needed to obtain a mutation rate matrix. Each position of the reference genome (NC_045512.2/MN908947.3/Wuhan-Hu-1; https://www.ncbi.nlm.nih.gov/nuccore/MN908947) was classified as coding or noncoding. Start and stop codons were not considered and were simulated as constant. The first and last 100 base pairs of the genome and sites marked as problematic in https://github.com/W-L/ProblematicSites_SARS-CoV2/blob/master/problematic_sites_sarsCov2.vcf were also not considered for estimating substitution rates but they were simulated like the other ‘normal’ sites. We counted ‘opportunities’ of mutations based on the reference genome: for example, a noncoding C allele in the reference genome represents three opportunities for noncoding mutations (C>A, C>G and C>U). For coding sites, we split synonymous and nonsynonymous mutation opportunities into two separate counts. Then, we counted the number of observed mutation events of each type using the inferred mutational history of the viral population based on the phylogeny and global alignment from 11 July 2020 and using the software we described previously (https://github.com/yatisht/strain_phylogenetics37). As above, we masked the ends of the genome, previously identified problematic sites and start/stop codons.
To further avoid potential bias resulting from sequencing errors, RNA degradation, contamination or other possible sources of error, we used a threshold for minor allele counts for variants/mutations to be included in our analysis. For all the results presented, we used rate estimates obtained including only variants/mutations with at least a minor allele count of 7. Results varied a little by varying this threshold; for example, the nonsynonymous/synonymous substitution rate ratio ω usually tended to decrease with increasing this threshold (ω = 0.48 with threshold 3, ω = 0.48 with threshold 7 and ω = 0.43 with threshold 20). However, overall, the effect is limited (Supplementary Tables 5–10). Also, we used two different ways to count mutation events. In the first approach, we used the reconstructed mutation histories from the strain_phylogenetics package (commit eb978ac) and only counted substitution events with at least seven descendant lineages. The other approach only used the alignment and ignored the reconstructed mutation history: we counted variant alleles in the alignment that had a minor allele count of at least seven. These two approaches give comparable results (Supplementary Tables 5–10); in this study, we used the results from the first approach.
For each class of mutations (synonymous or nonsynonymous) and from any nucleotide to any other nucleotide, we calculated a rate by dividing its total opportunity count by its mutation count. Given the small number of noncoding sites, we merged the noncoding and synonymous counts.
Simulating sequence evolution along the tree
We assumed a root genome equal to the reference genome and simulated its evolution along a phylogeny with 38,342 leaves using pyvolve v.1.0.1 (ref. 42). We simulated the evolution of each site with a nucleotide substitution process while still taking into account the genetic code and a non-neutral nonsynonymous/synonymous rate ratio (ω) in doing so. We achieved this by assuming that the codon context of each position was constant; this is sufficiently realistic because it is very rare that two positions in the same codon are both mutated (with respect to the reference) in the same SARS-CoV-2 sequence. So, considering each genome position in turn, we evolved noncoding sites under the synonymous matrix above; for coding sites, we used the same rates for synonymous mutations, while we multiplied the rate of nonsynonymous mutations by ω. The reason for using this approach instead of simulating under a codon model is that a codon model would have been prohibitively computationally intensive.
We assumed that ω was variable across sites and tested the resilience of our approach to variation in rates across sites. We chose a distribution of ω such that the average simulated ω was close to the ω inferred from real data: 20% probability of ω = 0.01; 25% probability of ω = 0.1; 25% probability of ω = 0.4; and 30% probability of ω = 1. Pyvolve automatically rescaled the input rate matrices, so we rescaled the tree at each site so that the synonymous rates were the same across sites. In addition, we rescaled the overall tree so that the observed number of mutation events in the simulated alignment was similar to the number in the real alignment.
Accuracy evaluation
We measured UShER’s accuracy in placing samples onto a reference phylogeny using simulated (described above) and real data. For simulated data, both reference phylogeny and sequences were simulated; for real data, we used the global phylogeny dated 11 July 2020 (https://github.com/roblanf/sarscov2phylo; Supplementary Data 1) as reference and its corresponding sequences were obtained from GISAID4. In each case, we first randomly pruned out ten samples from the global phylogeny, which was then used as the input phylogeny while adding back the pruned samples using UShER. UShER’s accuracy in placing back the samples was computed using the average values of three different statistics (described below) over 100 such replicates.
We initially used TreeCmp43 v.2.0-b76 to compute the Robinson–Foulds distance between the reference phylogeny and the tree constructed by samples using UShER. Separately, we recorded whether the sister clade for each placed sample was identical to the true sister clade (that is, the sister clade in the reference phylogeny). Finally, we computed the distance between the UShER placement and the correct placement in terms of the minimum number of edges separating them as described below.
Ordinarily, the distance between two nodes in a tree can be computed using their lowest common ancestor44 by taking the sum of the number of edges to each node from the lowest common ancestor. To determine the distance between the node placement in two trees (reference phylogeny and the one resulting from UShER placement), we developed a utility that reports all descendant lineages from an n-th generation ancestor of any given node in a tree, with n provided as input (that is, when n = 1, it reports unpruned lineages in the sister clade). For each pruned lineage, we found the descendants varying the number of generations, N1 and N2, in global and UShER phylogenies, respectively and reported the distance between the UShER placement and the correct placement as the smallest (N1 + N2 − 2), which resulted in the same set of descendant lineages in both phylogenies. Note that the second statistic records cases for which the sister clades in the two trees are identical, which would always have 0 distance in our third statistic (N1 = N2 = 1; Extended Data Fig. 6).
We also measured UShER’s accuracy in a more realistic scenario of placing closely related samples that form their own subtree. In this case, during pruning, we required that the pruned samples together formed a subtree (that is, not a trivial polytomy) in the reference phylogeny.
To evaluate the accuracy and robustness to error of UShER compared to IQ-TREE 2 and FastTree 2, we identified 5 clades of approximately 1,000 lineages and reconstructed each from scratch using each of the 3 methods, and repeated these experiments after randomly masking between 2.5 and 50% of sites to ‘n’, adding 10, 20 … 100 independently drawn random single-nucleotide errors across the lineages to be placed and adding identical single-nucleotide errors to 1–10 of the genomes to be placed (Extended Data Fig. 1). We measured the accuracy of these placements by calculating the Robinson–Foulds distance using TreeCmp43 v2.0-b76 (Extended Data Fig. 6).
Benchmarking placement algorithms
We compared UShER to four other lineage placement algorithms: IQ-TREE multicore v.2.1.1; EPA-ng v.0.3.8; PAGAN2 v.1.54; and TreeBEsT v.1.9.2 (refs. 17,18,19,20). We initially attempted to add 1,000 lineages to our simulated phylogeny; however, except or UShER, which required 18 s to finish using 64 threads, none of the placement programs finished within 24 h. Due to time and memory constraints, we instead added only 1 lineage to the tree in 20 replicates, recording the average and time range and peak memory usage across these 20 replicates in Table 1. A full list of commands used to run each test can be found in Supplementary Table 11. We installed and ran each program on a server with 160 processors (Intel Xeon CPU E7-8870 v.4, 2.10 gigahertz), each with 20 CPU cores.
Tree construction for SARS-CoV-2 samples
Full details and reproducible code for the construction of the global tree of SARS-CoV-2 samples are available in the 31 July 2020 release and at Lanfear22. To summarize, this code creates a global phylogeny of all available samples from the GISAID data repository as follows. First, all sequences marked as ‘complete’ and ‘high coverage’ submitted up to 31 July 2020 were downloaded from GISAID. Sequences with known issues from previous analyses were then removed from this database (details are in the excluded_sequences.tsv file; https://github.com/roblanf/sarscov2phylo/blob/master/excluded_sequences.tsv). Second, a global alignment was created by aligning every sequence individually to the NC_045512.2 accession from the National Center for Biotechnology Information using mafft v.7.471 (ref.45), faSplit (http://hgdownload.soe.ucsc.edu/admin/exe/), faSomeRecords (https://github.com/ENCODE-DCC/kentUtils), both version v.377, and GNU Parallel v.2020082246. This approach aligns each sequence individually to the reference, then joins them into a global alignment by ignoring insertions relative to the reference. Third, sites that are likely to be dominated by sequencing errors36 are masked from the alignment using faSplit, seqmagick v.0.7.0+7.g1642bb8 (https://seqmagick.readthedocs.io/en/latest/) and GNU Parallel; sequences shorter than 28 kilobases and/or with >1,000 ambiguities are removed from the alignment using esl-alimanip v.0.47 (http://www.hmmer.org/); subsequently, sites that have >50% gaps are removed (after converting N to gaps) with esl-alimask. Fourth, the global phylogeny is estimated using FastTree 2 (ref. 38) (v.2.1.10 compiled with double precision) in two stages: (1) an initial analysis that produces a neighbor joining tree, which is optimized with 5 rounds of subtree pruning and regrafting (SPR) moves of length 500; and (2) a second analysis, which uses the tree from the first analysis as a starting tree with 5 rounds of SPR moves of length 200 and otherwise default FastTree 2 settings. Finally, goalign (commit v.0.3.1; https://github.com/evolbioinfo/goalign) was used to create 100 bootstrap alignments followed by reestimating all the maximum-likelihood trees with FastTree 2 with the -fastest setting, using GNU Parallel to manage parallelization. The resulting trees were rooted with our reference (NC_045512.2/MN908947.3/Wuhan-Hu-1) sequence using nw_reroot v.1.6 (ref. 47).
From the resulting tree, we removed sequences on very long branches using TreeShrink v.1.3.7 (refs. 48,49) using the default q ≤ 0.05 threshold to identify such branches. These sequences are likely to be either of poor quality and/or poorly aligned, so rather unreliable to interpret in a phylogeny with such limited variation.
Tree optimization for inferred SARS-CoV-2 phylogenies
For the 11 and 31 July 2020 reference trees, we created ‘optimized’ versions of each using FastTree 2 (ref. 38) using ten iterations of the command ‘fasttree -nt -nni 0 -spr 1 -sprlength 1000 -nosupport -intree <initial tree> global.fa > <new tree>’, replacing the initial tree with the new tree from the previous iteration each time, followed by the command ‘fasttree -nt -nni 0 -spr 1 -sprlength 1000 -nosupport -gamma -intree <initial tree> global.fa > <new tree>’. In these commands, -nt indicates that the input is a nucleotide alignment, -nni 0 indicates that no minimum-evolution nearest neighbor interchanges are done, -spr 1 -sprlength 1000 indicates 1 round of SPR with a maximum distance of 1,000, meaning that a single SPR move could move a subtree to any new branch of the global tree. The -nosupport flag indicates that support values are not output and the -gamma flag indicates that the lengths are rescaled to optimize the Gamma20 likelihood38. Because FastTree 2 requires binary trees, we randomly resolved all polytomies before optimization. We also generated two other trees using UShER, by taking the original and ‘optimized’ 11 July trees, pruning out all lineages in the 11 July 2020 tree not present in the 31 July 2020 tree and using UShER to add in all lineages present in the 31 July 2020 that were not present in the 11 July tree. We then optimized these two new trees using ten iterations of FastTree 2, followed by another round of optimization using the -gamma flag as described above.
Parsing the mutation-annotated tree for subtree visualization
When we were developing the Web application for UShER, we discovered that parsing genotype data from a VCF file containing 40,000+ samples was the most time-consuming step for displaying the resulting subtrees. Therefore, we developed an approach for parsing genotype data from subtrees from the mutation-annotated tree object. Briefly, our approach descends from the root of the phylogeny to the focal subtree, accumulates the relevant mutations along the path and then extracts the variation within the subtree that will be used for visualization. This heavily reduced dataset can then be visualized using the existing code base of the UCSC Genome Browser, outputting a JSON-formatted file that can be viewed using Auspice (https://nextstrain.github.io/auspice/). With the current dataset sizes, this procedure takes approximately 0.03 s in total to extract genotype data for a subtree of 50 sequences. Software for rapid subtree VCF extraction from our mutation-annotated tree object is available at https://github.com/ucscGenomeBrowser/kent/tree/master/src/hg/hgPhyloPlace/phyloPlace.c.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data used in this work are available from GISAID (https://www.gisaid.org/), with specific sample accessions listed in Supplementary Data 2–6.
Code availability
UShER is available to users through the UCSC Genome Browser at https://genome.ucsc.edu/cgi-bin/hgPhyloPlace. The source code and detailed instructions on how to compile and run UShER are available at https://github.com/yatisht/usher. The code used for the statistical analyses and to produce the figures is available at https://github.com/bpt26/USHER_ANALYSES.
References
Lam, T. T.-Y. et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 583, 282–285 (2020).
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C. & Garry, R. F. The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Shu, Y. & McCauley, J. GISAID: global initiative on sharing all influenza data—from vision to reality. Euro Surveill. 22, 30494 (2017).
Stefanelli, P. et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: additional clues on multiple introductions and further circulation in Europe. Euro Surveill. 25, 2000305 (2020).
Surleac, M. et al. Molecular epidemiology analysis of SARS-CoV-2 strains circulating in Romania during the first months of the pandemic. Life (Basel) 10, 152 (2020).
Deng, X. et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 369, 582–587 (2020).
Pattabiraman, C. et al. Genomic epidemiology reveals multiple introductions and spread of SARS-CoV-2 in the Indian state of Karnataka. PLoS ONE 15, e0243412 (2020).
Maurano, M. T. et al. Sequencing identifies multiple early introductions of SARS-CoV-2 to the New York City region. Genome Res. 30, 1781–1788 (2020).
Gámbaro, F. et al. Introductions and early spread of SARS-CoV-2 in France, 24 January to 23 March 2020. Euro Surveill. 25, 2001200 (2020).
Thielen, P. M. et al. Genomic diversity of SARS-CoV-2 during early introduction into the United States National Capital Region. Preprint at medRxiv https://doi.org/10.1101/2020.08.13.20174136 (2020).
Rockett, R. J. et al. Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 26, 1398–1404 (2020).
Dellicour, S. et al. A phylodynamic workflow to rapidly gain insights into the dispersal history and dynamics of SARS-CoV-2 lineages. Mol. Biol. Evol. 38, 1608–1613 (2021).
Fauver, J. R. et al. Coast-to-coast spread of SARS-CoV-2 during the early epidemic in the United States. Cell 181, 990–996.e5 (2020).
Lu, J. et al. Genomic epidemiology of SARS-CoV-2 in Guangdong province, China. Cell 181, 997–1003.e9 (2020).
Bedford, T. et al. Cryptic transmission of SARS-CoV-2 in Washington State. Science 370, 571–575 (2020).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Barbera, P. et al. EPA-ng: massively parallel evolutionary placement of genetic sequences. Syst. Biol. 68, 365–369 (2019).
Löytynoja, A., Vilella, A. J. & Goldman, N. Accurate extension of multiple sequence alignments using a phylogeny-aware graph algorithm. Bioinformatics 28, 1684–1691 (2012).
Ruan, J. et al. TreeFam: 2008 update. Nucleic Acids Res. 36, D735–D740 (2008).
Singer, J., Gifford, R., Cotten, M. & Robertson, D. CoV-GLUE: a web application for tracking SARS-CoV-2 genomic variation. Preprint at Preprints.org https://doi.org/10.20944/preprints202006.0225.v1 (2020).
Lanfear, Robert. A global phylogeny of SARS-CoV-2 sequences from GISAID. Zenodo https://doi.org/10.5281/zenodo.3958883 (2020).
Simon, C. An evolving view of phylogenetic support. Syst. Biol. https://doi.org/10.1093/sysbio/syaa068 (2020).
Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 (1985).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Anisimova, M. & Gascuel, O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552 (2006).
Fernandes, J. D. et al. The UCSC SARS-CoV-2 Genome Browser. Nat. Genet. 52, 991–998 (2020).
Fitch, W. M. Toward defining the course of evolution: minimum change for a specific tree topology. Syst. Zool. 20, 406–416 (1971).
Sankoff, D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 28, 35–42 (1975).
Ralph, P., Thornton, K. & Kelleher, J. Efficiently summarizing relationships in large samples: a general duality between statistics of genealogies and genomes. Genetics 215, 779–797 (2020).
Kelleher, J., Thornton, K. R., Ashander, J. & Ralph, P. L. Efficient pedigree recording for fast population genetics simulation. PLoS Comput. Biol. 14, e1006581 (2018).
Hennessy, J. L. & Patterson, D. A. Computer Architecture: A Quantitative Approach (Elsevier, 2017).
Felsenstein, J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Zool. 27, 401–410 (1978).
Morel, B. et al. Phylogenetic analysis of SARS-CoV-2 data is difficult. Mol. Biol. Evol. https://doi.org/10.1093/molbev/msaa314 (2020).
Turakhia, Y. et al. Stability of SARS-CoV-2 phylogenies. PLoS Genet. 16, e1009175 (2020).
De Maio, N. et al. Issues with SARS-CoV-2 sequencing data. Preprint at https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Auspice v.2.0 (Nextstrain, 2020).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Adding Extra Metadata via CSV/TSV—Auspice Documentation. https://docs.nextstrain.org/projects/auspice/en/latest/advanced-functionality/drag-drop-csv-tsv.html (2020).
Spielman, S. J. & Wilke, C. O. Pyvolve: a flexible Python module for simulating sequences along phylogenies. PLoS ONE 10, e0139047 (2015).
Bogdanowicz, D., Giaro, K. & Wróbel, B. TreeCmp: comparison of trees in polynomial time. Evol. Bioinform. Online 8, 475–487 (2012).
Bender, M. A., Farach-Colton, M., Pemmasani, G., Skiena, S. & Sumazin, P. Lowest common ancestors in trees and directed acyclic graphs. J. Algorithms 57, 75–94 (2005).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Tange, O. GNU Parallel: the command-line power tool. USENIX Mag. https://www.usenix.org/publications/login/february-2011-volume-36-number-1/gnu-parallel-command-line-power-tool (2011)..
Junier, T. & Zdobnov, E. M. The Newick utilities: high-throughput phylogenetic tree processing in the Unix shell. Bioinformatics 26, 1669–1670 (2010).
Mai, U. & Mirarab, S. TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genomics 19, 272 (2018).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Robinson, D. F. & Foulds, L. R. in Combinatorial Mathematics VI (eds Horadam, A. F. & Wallis, W. D.) 119–126 (1979).
Bouckaert, R. R. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26, 1372–1373 (2010).
Acknowledgements
We thank all of the Nextstrain team members whose platform we have built on as a part of this work. We also thank J. Batson, S. Bell and M. Bui for providing feedback on the UShER features. Until about the 24 June 2020, GISAID acknowledgements were provided in tabulated form on their website; therefore, they could be combined into a single file. This is Supplementary Data 2. Subsequent to this, GISAID shifted to only allowing PDF downloads of this table. However, it is not possible to download the entire table at once, only small subsections of it. As a result, acknowledgements after the 24 June 2020 are provided in PDF format (Supplementary Data 3–6). This statement applies to all formats of the acknowledgement tables. We thank the following authors from the originating laboratories responsible for obtaining the specimens and the submitting laboratories where genome data were generated and shared via GISAID, on which this research is based. During this work, Y.T is funded through Schmidt Futures Foundation SF 857 and NIH grant 5R01HG010485. B.T. and R.C.-D. were supported by grant no. R35GM128932 and by an Alfred P. Sloan Foundation Fellowship to R.C.-D. B.T. was funded by grant nos. T32HG008345 and F31HG010584. The UCSC Human Genome Browser software, quality control, and training is funded by National Human Genome Research Institute, currently with grant no. 5U41HG002371-19. The SARS-CoV-2 genome browser and data annotation tracks are funded by generous individual donors including P. and R. Rebele, E. and W. Schmidt by recommendation of the Schmidt Futures program, the Center for Information Technology Research in the Interest of Society (no. 2020-0000000020) and a University of California Office of the President Emergency COVID-19 Research Seed Funding Grant no. R00RG2456. N.D.M. is funded by the European Molecular Biology Laboratory. R.L. is funded by an Australian Research Council grant no. DP200103151 and by a Chan Zuckerberg Initiative grant.
Author information
Authors and Affiliations
Contributions
R.C.-D. and Y.T. conceived and designed the research. N.D.M., B.T., A.S.H., N.D.M., L.G. and R.L. performed the research and analyses. Y.T., A.S.H. and L.G. developed the software tools. D.H. and R.C.-D. contributed reagents and resources. R.C.-D., Y.T. and B.T. wrote the manuscript with input from all authors. All authors edited and contributed to the manuscript revision.
Corresponding authors
Ethics declarations
Competing interests
A.S.H. and D.H. receive royalties from the sale of UCSC Genome Browser source code licenses to commercial entities. R.L. works as an advisor to GISAID. The remaining authors declare no competing interests.
Additional information
Peer review information Nature Genetics thanks Matthew Hall and Art Poon for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 UShER is similarly robust to masked sites and nucleotide errors as IQ-TREE2 and FastTree 2.
We pruned 5 independent clades of roughly 1,000 lineages each and applied the same methods as in Fig. 2, masking 2.5, 5, 7.5, 10, 20...50 percent of sites (top, note that the X-axis does not use a linear scale), adding 10, 20...100 independently drawn random nucleotide substitutions across the lineages to be placed (center), and adding one error to 1, 2...10 of the genomes of interest (bottom). We then used UShER (blue), IQ-TREE 217 (orange), and FastTree 238 (purple) to reconstruct these clades. We determined the Robinson-Foulds distance of each to the original clade using TreeCmp43, as well as the distance of randomly constructed trees to the far right (black, labeled ‘Null’) as a null model comparison. N = 5 independent replicates for each experiment. Each boxplot is centered on the median of the data and extends to the first and third quartiles, with whiskers extending to the minimum and maximum of the data set.
Extended Data Fig. 2 Addition of two perfectly correlated errors significantly reduces UShER accuracy.
As in Fig. 2, the Robinson-Foulds distances, proportion of sister nodes identical to the reference tree, distance from true placement and equally parsimonious placements, respecitvely, are shown for UShER experiments in placing 10 lineages, with two perfectly correlated errors added to 1, 2 … 10 of the lineages to be placed. To the far right in the left-most panel, labeled ‘Null’, the distribution of scores across 100 replicates in which 10 lineages were added randomly to the phylogeny is shown as a null model for comparison. N = 100 independent replicates for each experiment. The whiskers in the boxplot on the left are centered on the median of the data and extend to the first and third quartiles. In the error bars panel (second from the left), the data points are centered on the mean of the data and extend to the bounds of the 95% confidence interval, calculated by 1,000 iterations of bootstrapping.
Extended Data Fig. 3 UShER can output multiple trees to accommodate phylogenetic uncertainty.
(A): Composite of 239 trees with 424 samples, representing all possible parsimony-optimal placements of two samples on a starting tree having 422 samples, computed using DensiTree52 and plotted using the phangorn package (https://cran.r-project.org/web/packages/phangorn). All trees were scaled to be the same height. (B): Two of the trees from (A) compared in a tanglegram, colored according to COG-UK lineage assignments, with linker lines shown only for the two placed samples whose placements differ between topologies. As in Fig. 4, both trees in this tanglegram are ultrametric and branch lengths are arbitrary.
Extended Data Fig. 4 UCSC Genome Browser display of subtree where hypothetical example sequences have been placed by UShER.
Newly added samples are highlighted in blue and the tree displaying their relationships and placement on the global tree is shown to the left. Interactive view: https://genome.ucsc.edu/s/AngieHinrichs/UShER_example.
Extended Data Fig. 5 Nextstrain/Auspice view of subtree created by UShER placing the same hypothetical example samples.
As in Extended Data Fig. 4. Direct link: https://nextstrain.org/fetch/hgwdev.gi.ucsc.edu/~angie/usher_example.json.
Extended Data Fig. 6 A demonstration of our distance metric for placements.
To evaluate the accuracy of each placement in a new phylogeny, we compute the distance for each newly placed sample in the UShER tree (Tree 1) with the reference tree (Tree 2). The clade sets in the two trees are shown for each N1 and N2 value, representing the number of generations from the Sample D in Tree 1 and Tree 2, respectively. We compute the values of N1+N2-2 such that the descendant clades for both trees are identical. In case of newly placed Sample D, clades are identical when N1=2 and N2=2 and when N1=3 and N2=3, which are highlighted in bold. Hence the distance (smallest N1+N2-2) from the true placement is equal to 2.
Supplementary information
Supplementary Tables
Supplementary Tables 1–11
Supplementary Data 1
Newick-formatted trees created in this work. In order, the phylogenies in this file are (1) the reference 31 July 2020 tree, (2) the optimized 31 July 2020 tree, (3) the 11 July 2020 reference tree with missing samples added by UShER, (4) the 11 July 2020 optimized tree with samples added by UShER, (5) the 11 July 2020 reference tree with samples added by UShER and then optimized, (6) the 11 July 2020 optimized tree with samples added by UShER then optimized again, (7) the 11 July 2020 reference tree with missing samples added at random and (8) the 11 July 2020 optimized tree with missing samples added at random.
Supplementary Data 2
GISAID data providers acknowledgement table.
Supplementary Data 3
GISAID data providers acknowledgement table.
Supplementary Data 4
GISAID data providers acknowledgement table.
Supplementary Data 5
GISAID data providers acknowledgement table.
Supplementary Data 6
GISAID data providers acknowledgement table.
Rights and permissions
About this article

Cite this article
Turakhia, Y., Thornlow, B., Hinrichs, A.S. et al. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat Genet 53, 809–816 (2021). https://doi.org/10.1038/s41588-021-00862-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00862-7
This article is cited by
-
A framework for automated scalable designation of viral pathogen lineages from genomic data
Nature Microbiology (2024)
-
Using big sequencing data to identify chronic SARS-Coronavirus-2 infections
Nature Communications (2024)
-
The ongoing evolution of UShER during the SARS-CoV-2 pandemic
Nature Genetics (2024)
-
Positive selection underlies repeated knockout of ORF8 in SARS-CoV-2 evolution
Nature Communications (2024)
-
Randomized controlled trial of molnupiravir SARS-CoV-2 viral and antibody response in at-risk adult outpatients
Nature Communications (2024)
