
Abstract
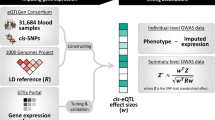
Here, we present a joint-tissue imputation (JTI) approach and a Mendelian randomization framework for causal inference, MR-JTI. JTI borrows information across transcriptomes of different tissues, leveraging shared genetic regulation, to improve prediction performance in a tissue-dependent manner. Notably, JTI includes the single-tissue imputation method PrediXcan as a special case and outperforms other single-tissue approaches (the Bayesian sparse linear mixed model and Dirichlet process regression). MR-JTI models variant-level heterogeneity (primarily due to horizontal pleiotropy, addressing a major challenge of transcriptome-wide association study interpretation) and performs causal inference with type I error control. We make explicit the connection between the genetic architecture of gene expression and of complex traits and the suitability of Mendelian randomization as a causal inference strategy for transcriptome-wide association studies. We provide a resource of imputation models generated from GTEx and PsychENCODE panels. Analysis of biobanks and meta-analysis data, and extensive simulations show substantially improved statistical power, replication and causal mapping rate for JTI relative to existing approaches.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
The protected data for the GTEx project (for example, genotype and RNA sequencing data) are available via access request to the database of Genotypes and Phenotypes (accession number phs000424.v8.p2). Processed GTEx data (for example, gene expression and eQTLs) are available from the GTEx portal (https://gtexportal.org). The URLs of the summary statistics datasets of all of the GWAS meta-analyses analyzed in this paper can be found in Supplementary Table 7. All of the summary results from the gene-based analyses are provided in Supplementary Table 7. The JTI GTEx models (as well as the PrediXcan and (modified) UTMOST models we generated) are available for download from Zenodo (https://doi.org/10.5281/zenodo.3842289). The PsychENCODE (https://doi.org/10.5281/zenodo.3859065) and GEUVADIS (https://doi.org/10.5281/zenodo.3859075) models have also been deposited.
Code availability
The code for JTI and MR-JTI and for reproducing the figures in this paper is available from GitHub (https://github.com/gamazonlab/MR-JTI).
References
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Barbeira, A. N. et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51, 568–576 (2019).
Flutre, T., Wen, X., Pritchard, J. & Stephens, M. A statistical framework for joint eQTL analysis inmultiple tissues. PLoS genetics 9 (2013).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Urbut, S. M., Wang, G., Carbonetto, P. & Stephens, M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 51, 187–195 (2019).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Pierce, B. L. & Burgess, S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178, 1177–1184 (2013).
Burgess, S. & Thompson, S. G. Use of allele scores as instrumental variables for Mendelian randomization. Int. J. Epidemiol. 42, 1134–1144 (2013).
Smith, G. D. & Ebrahim, S. Mendelian randomization: prospects, potentials, and limitations. Int. J. Epidemiol. 33, 30–42 (2004).
Johnson, T. Efficient Calculation for Multi-SNP Genetic Risk Scores Technical Report (The Comprehensive R Archive Network, 2013).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J. et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Verbanck, M., Chen, C.-Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Nagpal, S. et al. Tigar: an improved Bayesian tool for transcriptomic data imputation enhances gene mapping of complex traits. Am. J. Hum. Genet. 105, 258–266 (2019).
Barbeira, A. N. et al. Widespread dose-dependent effects of RNA expression and splicing on complex diseases and traits. Preprint at BioRxiv https://doi.org/10.1101/814350 (2019).
Bavoux, C., Hoffmann, J. & Cazaux, C. Adaptation to DNA damage and stimulation of genetic instability: the double-edged sword mammalian DNA polymerase κ. Biochimie 87, 637–646 (2005).
Williams, H. L., Gottesman, M. E. & Gautier, J. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell 47, 140–147 (2012).
Shimizu, I., Yoshida, Y., Suda, M. & Minamino, T. DNA damage response and metabolic disease. Cell Metab. 20, 967–977 (2014).
Stancel, J. N. K. et al. Polk mutant mice have a spontaneous mutator phenotype. DNA Repair 8, 1355–1362 (2009).
Li, Z. et al. Integrating mouse and human genetic data to move beyond GWAS and identify causal genes in cholesterol metabolism. Cell Metab. 31, 741–754.e5 (2020).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390.e19 (2019).
Gamazon, E. R. et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 50, 956–967 (2018).
Gratten, J. & Visscher, P. M. Genetic pleiotropy in complex traits and diseases: implications for genomic medicine. Genome Med. 8, 78 (2016).
Mancuso, N. et al. Probabilistic fine-mapping of transcriptome-wide association studies. Nat. Genet. 51, 675–682 (2019).
Wen, X., Pique-Regi, R. & Luca, F. Integrating molecular QTL data into genome-wide genetic association analysis: probabilistic assessment of enrichment and colocalization. PLoS Genet. 13, e1006646 (2017).
ENCODE Project Consortium The ENCODE (Encyclopedia of DNA Elements) project. Science 306, 636–640 (2004).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Barfield, R. et al. Transcriptome-wide association studies accounting for colocalization using Egger regression. Genet. Epidemiol. 42, 418–433 (2018).
Wainberg, M. et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 51, 592–599 (2019).
Deschenes, A. et al. similaRpeak: Metrics to estimate a level of similarity between two ChIP-Seq profiles. R package version 1.18.0 (2019).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018).
Teslovich, T. M. et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713 (2010).
Roden, D. M. et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 84, 362–369 (2008).
Knight, K. & Fu, W. Asymptotics for lasso-type estimators. Ann. Stat. 28, 1356–1378 (2000).
Chatterjee, A. & Lahiri, S. Asymptotic properties of the residual bootstrap for lasso estimators. Proc. Am. Math. Soc. 138, 4497–4509 (2010).
Chatterjee, A. & Lahiri, S. N. Bootstrapping lasso estimators. J. Am. Stat. Assoc. 106, 608–625 (2011).
Burgess, S. et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 4, 186 (2019).
Hartwig, F. P., Davies, N. M., Hemani, G. & Davey Smith, G. Two-Sample Mendelian Randomization: Avoiding the Downsides of a Powerful, Widely Applicable but Potentially Fallible Technique (Oxford Univ. Press, 2016).
Acknowledgements
E.R.G. is grateful to the president and fellows of Clare Hall, University of Cambridge for providing a stimulating intellectual home and for generous support during the Lent and Easter terms (2018). E.R.G. is supported by the National Human Genome Research Institute of the National Institutes of Health under award numbers R35HG010718 and R01HG011138. N.J.C. is supported by U01HG009086 and R01MH113362. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The datasets used for part of the replication analysis were obtained from Vanderbilt University Medical Center’s BioVU biorepository, which is supported by institutional funding, private agencies and federal grants, including the NIH-funded Shared Instrumentation Grant S10RR025141 and Clinical and Translational Science Award grants UL1TR002243, UL1TR000445 and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962 and R01HD074711, as well as the additional funding sources listed at https://victr.vanderbilt.edu/pub/biovu/. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS.
Author information
Authors and Affiliations
Contributions
E.R.G. and D.Z. designed the study and wrote the manuscript. E.R.G. and D.Z. developed and implemented the methodology. D.Z. performed the analyses. Y.J., X.Z., N.J.C. and C.L. provided critical input and contributed to reviewing and editing the manuscript. E.R.G. supervised and acquired funding for the study.
Corresponding authors
Ethics declarations
Competing interests
E.R.G. receives an honorarium from the journal Circulation Research of the American Heart Association as a member of the Editorial Board. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 TWAS could be biased by possible sources of bias (PSB), including invalid instrumental variables (IVs) due to horizontal pleiotropy and weak instruments.
Conventional TWAS, such as PrediXcan, can be viewed as Mendelian randomization with multiple IVs, but without horizontal pleiotropy control. a, A major source of false positives from TWAS is the use of invalid IVs due to horizontal pleiotropy. b, Horizontal pleiotropy can arise in multiple ways. For example, it can come from LD-induced invalid IVs, that is, LD contamination. If we are testing the significance of Gene A, but one of the SNPs (yellow) in the prediction model tags another coding (red) or regulatory (blue) variant that is causal for the trait through another Gene B, causal effect estimation will be biased. c, Even without LD contamination, the estimation may also be biased by the inclusion of weak or false positive eQTLs in the prediction model for Gene A. In this case, the effect of the weak or false positive eQTL for Gene A on the trait is actually mediated by another Gene B (by affecting coding or regulation). More generally, weak instrument bias is a type of finite sample bias; it arises in finite samples where the gene expression (“exposure”) is only weakly correlated with the instrument set. Both b, and c, result in d, a biased estimate of gene causal effect on trait. We estimate the heterogeneity due to PSB using threshold-based residual bootstrap LASSO (see Methods). Our approach estimates the heterogeneity due to invalid IVs and gives an adjusted estimate of the gene causal effect on trait.
Extended Data Fig. 2 The gene expression similarity matrix.
The median expression level (log2-transformed TPM) across all the samples of a given tissue was used to evaluate the correlation (Pearson) of tissue-tissue pairs across the transcriptome. The similarity map was generated by performing hierarchical clustering.
Extended Data Fig. 3 Comparison of prediction performance between PrediXcan and JTI in all GTEx v8 tissues.
We compared the performance of PrediXcan and JTI using the Pearson correlation r between predicted and observed expression levels for each of the 49 GTEx v8 tissues with more than 70 samples. The white box edges depict interquartile range, whiskers 1.5× the interquartile range, center black dot marks the median level, and the outlines display the kernel probability density. The median correlation is also shown below the x-axis label.
Extended Data Fig. 4 Prediction performance comparison between JTI and three single-tissue approaches (top eQTL, BSLMM, and DPR) in two independent datasets.
Prediction models were trained using BLSMM (5-fold cross-validation FUSION default setting) and JTI (see Methods) in GTEx v8 a, brain frontal cortex BA9 region and d, EBV-transformed lymphocytes. The x-axis and y-axis represent the Pearson correlation r between the predicted expression and observed expression in external (non-GTEx and independent) datasets, that is, a, PsychENCODE and d, GEUVADIS. b, and e, show the corresponding comparisons between JTI and top eQTL, which simply models the genetically regulated expression using the top eQTL. c, and f, We also compared the prediction performance with the DPR model, a nonparametric Bayesian method with a Dirichlet process prior on effect-size variance, using the software tool ‘TIGAR’ with 5-fold cross-validation. The green and purple circles denote genes imputable using only JTI and the other single-tissue approaches, respectively. The black and grey dots denote genes consistently imputable and not imputable, respectively, using both methods.
Extended Data Fig. 5 JTI and PrediXcan showed a substantial increase in iGene discovery between GTEx v6p and v8.
We compared the number of imputable genes across all the tissues between GTEx v6p (yellow) and v8 (green). The prediction performance of GTEx v8 was superior to v6p for both a, PrediXcan and b, JTI in all tissues. The number of iGenes can be found in Supplementary Table 1.
Extended Data Fig. 6 Prediction performance comparison among PrediXcan, JTI, original UTMOST, and modified UTMOST for brain frontal cortex BA9.
We compared the cross-validation prediction performance (r2) in GTEx (internal, brain frontal cortex BA9) and the prediction performance in PsychENCODE (an external test data set, brain prefrontal cortex) among a, PrediXcan, b, JTI, c, original UTMOST, and d, modified UTMOST. The lower figures e–h, are the zoom-in version of the corresponding upper figures. The yellow, green, and purple dots indicate high, medium, and low density.
Extended Data Fig. 7 Prediction performance comparison among PrediXcan, JTI, original UTMOST, and modified UTMOST for EBV transformed lymphocytes.
We compared the cross-validation prediction performance (r2) in GTEx (internal, EBV transformed lymphocytes) and the prediction performance in GEUVADIS (an external test data set, LCLs) among a, PrediXcan, b, JTI, c, original UTMOST, and d, modified UTMOST. The lower figures e, f, g, and h, are the zoom-in version of the corresponding upper figures. The yellow, green, and purple dots indicate high, medium, and low density.
Extended Data Fig. 8 Type I error rate for PrediXcan, UTMOST, and JTI.
The Q-Q plots show the type I error from applying PrediXcan, UTMOST, and JTI models in a–c brain frontal cortex BA9 and d–f, Liver. The blue dashed lines show the 95% CI of the expected -log(P). Type I error rate for all the tissues can be found in Supplementary Table 3.
Extended Data Fig. 9 TWAS power analysis for PrediXcan, UTMOST, and JTI.
The true expression level of randomly sampled causal genes and the effect size for each gene on trait were simulated. In this model, each gene, on average, contributed 0.5% to the phenotypic variance. For each gene, the predicted (that is, genetically determined) expression level was generated according to the proportion of variance explained (PVE), based on the actual prediction performance (R2) in two external datasets (a, PsychENCODE and b, GEUVADIS), for each of the three imputation approaches (PrediXcan, UTMOST, and JTI). Power was estimated as the proportion of simulations that attain significance (defined as Bonferroni adjusted P < 0.05).
Extended Data Fig. 10 Comparison of the estimated gene effect size on LDL-C from MR-JTI and the median estimator.
For each gene, the median estimator was calculated as the median of the Wald ratio estimates across all the cis-eQTLs. The Wald ratio estimate for a cis-eQTL is the ratio of the estimate for the GWAS effect size and the estimate for the eQTL effect size. a, Positive correlation between the estimated gene effect size from MR-JTI and the median estimator effect size (Spearman r = 0.72, P < 2.2e-16) was observed. b, No significant correlation was observed between the median estimator and the MR-JTI estimate from shuffled GWAS summary statistics data. Furthermore, note that MR-JTI’s type I error is well-controlled.
Supplementary information
Supplementary Information
Supplementary Note and Figs. 1–4
Supplementary Tables
Supplementary Tables 1–10
Rights and permissions
About this article

Cite this article
Zhou, D., Jiang, Y., Zhong, X. et al. A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nat Genet 52, 1239–1246 (2020). https://doi.org/10.1038/s41588-020-0706-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-020-0706-2
This article is cited by
-
Tissue-specific atlas of trans-models for gene regulation elucidates complex regulation patterns
BMC Genomics (2024)
-
Incorporating genetic similarity of auxiliary samples into eGene identification under the transfer learning framework
Journal of Translational Medicine (2024)
-
The broad impact of cell death genes on the human disease phenome
Cell Death & Disease (2024)
-
Adjusting for genetic confounders in transcriptome-wide association studies improves discovery of risk genes of complex traits
Nature Genetics (2024)
-
Investigating the role of common cis-regulatory variants in modifying penetrance of putatively damaging, inherited variants in severe neurodevelopmental disorders
Scientific Reports (2024)
